


Abstract
Chemical space is routinely explored by machine learning methods to discover interesting molecules, before time-consuming experimental synthesizing is attempted. However, these methods often rely on a graph representation, ignoring 3D information necessary for determining the stability of the molecules. We propose a reinforcement learning (RL) approach for generating molecules in Cartesian coordinates allowing for quantum chemical prediction of the stability. To improve sample-efficiency we learn basic chemical rules from imitation learning (IL) on the GDB-11 database to create an initial model applicable for all stoichiometries. We then deploy multiple copies of the model conditioned on a specific stoichiometry in a RL setting. The models correctly identify low energy molecules in the database and produce novel isomers not found in the training set. Finally, we apply the model to larger molecules to show how RL further refines the IL model in domains far from the training data.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Discovering novel molecules or materials with desirable properties is a challenging task because of the immense size of chemical compound space. Further complicating the process is the costly and time-consuming process of synthesizing and testing proposed structures. Whereas this procedure historically was driven by a trial-and-error process, the advance of computational quantum chemical methods allows for initial screening to select promising molecules for experimental testing. While the computational resource growth provided a significant speed-up in processing molecules an exhaustive search of molecular compound space is still infeasible. Instead an automated search for interesting candidates is desired. Examples of such search methods include evolutionary algorithms [1, 2], basin-hopping [3] and particle swarm optimization [4].
More recently, machine learning (ML) enhanced versions of aforementioned methods [5–7] and regression methods facilitating speed-up of virtual screening [8, 9] have gained considerable interest by making researchers able to quickly identify attractive candidates for experimental testing. An added benefit of virtual screening is the creation of numerous databases containing structures with computed chemical and physical properties [10–14] leading to generative models for discovery of novel molecules and materials [15–26]. Unlike virtual screening where candidates are selected among the structures in the database, generative models have shown a remarkable ability to leverage the database to produce new structures with desirable properties. A notable limitation is a large database and no feedback-loop to improve the generated molecules beyond what is learned from the database. To remedy this, reinforcement learning (RL) methods have started to become a competitive alternative [27–38] to methods relying on existing databases. RL involves a model that produces molecules and obtains properties for these molecules, by some external means other than a database. This provides the basis for the model to learn from the molecules it produces. Whereas generative models rely on databases for pretraining, RL is usually done without any prior knowledge leading to initial inefficiency as basic chemical and physical rules are learned. Instead a reward must be defined, meaning that it requires a setting where a meaningful reward function is available, such as an energy or other things to be optimized.
In this work we build upon a previous RL algorithm called Atomistic Structure Learning Algorithm (ASLA) [33] by incorporating databases into molecular RL to improve sample efficiency while simultaneously allowing the ML model to learn beyond the knowledge contained in the database. First, a general purpose model is trained using a database to create a model applicable for all stoichiometries. Then, a copy of the general purpose model is created for each stoichiometry of interest. These models are then further refined in a RL setting in a search for low energy isomers for the given stoichiometry. Specifically, we utilize a very small subset of the GDB-11 [39] database that consists of small organic molecules satisfying simple chemical stability rules and containing up to 11 atoms of C, N, O and F. We demonstrate that our RL model is able to both replicate structures in the database and produce novel low energy structures. By focusing on low energy structures, we believe we avoid the general pitfall of generative models that may produce a large degree of unsynthesizable molecules, as e.g. shown in the work of Gao and Connor [40]. Unlike previous approaches using SMILES [41] or graphs, we operate directly in Cartesian coordinates thus allowing for optimization of the potential energy hence easily biasing the search towards thermodynamically stable structures. To improve sample-efficiency we take a model-based approach where, similar to [36], the potential energy surface is modeled by a neural network allowing for approximate but cheap optimization. Finally, we introduce an architecture based on SchNet [42] with a self-attention mechanism [43] allowing the RL model to account for long-range effects. The paper is constructed as follows: first the RL theory is outlined, followed by a description of the neural network architecture. Then the database pretraining and RL phase are described before demonstrating the method on a subset of GDB-11. Finally, we apply the method to larger molecules outside the training distribution.
2. Theory
The objective of RL is to solve a decision problem, i.e. formulate a program that given an input can decide on the optimal action. Specifically, we refer to this program as an agent which is given a state, s, and must decide on an action, a. In the general case the decision process involves multiple steps indexed with the subscript t while T is used for the final step. In order to solve the problem the agent must device a policy, π, which is a probability distribution over the possible actions in a given state. The optimal policy is the policy which maximizes the sum of rewards, where r(s) is the reward given in state s. The states, actions and rewards are user-specified and describes the problem to be solved. Following [33, 34], the state space is defined as all (possibly partial) molecules along with a bag of atoms [34] not yet attached to the molecule. For the reward we seek to minimize the potential energy, i.e. we set intermediate rewards to zero and assign a final reward based on the potential energy of the molecule:

here Eref is the lowest energy observed and  is the energy span from Eref where energy differences are resolved. Note that
is the energy span from Eref where energy differences are resolved. Note that  meaning that
meaning that  . We scale the reward to stabilize the training as energies of produced structures may fluctuate substantially during the RL phase. We find that the steady lowering of Eref during searches caused by the identification of ever better structures constitutes a manageable learning setting. Since the reward function depends on the total energy of the molecule, separate agents must be trained for each stoichiometry of interest. This represents an appealing case for trivial parallelisation over the agents.
. We scale the reward to stabilize the training as energies of produced structures may fluctuate substantially during the RL phase. We find that the steady lowering of Eref during searches caused by the identification of ever better structures constitutes a manageable learning setting. Since the reward function depends on the total energy of the molecule, separate agents must be trained for each stoichiometry of interest. This represents an appealing case for trivial parallelisation over the agents.
To maximize the expected sum of rewards we define the Q-value,

i.e. the expected final reward when taking action a in state s and then following policy π. The goal of the agent is to infer the optimal Q-value function:

thereby enabling the agent to perform the best action in every state. To improve the Q-value function we parameterize it by a neural network and update it as the agent collects new experience. Specifically, we evaluate the Q-values on a voxelated grid [35], which allows us to update the Q-value towards the highest final reward observed by the agent for a specific state-action pair (figure 1), which for a deterministic problem puts a lower bound on the optimal Q-value [44]. Additionally, this discretization allows for easy inference of the optimal atom placement by simply finding the voxel which maximizes the Q-value.

Figure 1. Given a state, st and an action at , the target Q-value is calculated by computing the energy of finished structures involving the state-action pair. In the above case a hydrogen atom is placed, which has led to four different structures. The Q-value is updated towards the reward for the lowest energy structure built.
Download figure:
Standard image High-resolution image3. Architecture
The state representation is utilized for calculating the energy of a structure as well as a state value which is used for calculating the Q-value, but is independent of the specific action. For improved data efficiency the state and action representation must satisfy symmetries in the Hamiltonian, i.e. translation, rotation and mirroring of the molecule as well as permutation of identical atoms. To incorporate these properties in the state representation we utilize pairwise distances (figure 2(a), D), expanded in a Gaussian basis:

where γ = 1 Å−2 and rij
is the distance between atom i and j, with a total of n atoms in the structure. We let k be integer values given by  which corresponds to 20 values for µk
, uniformly between 0 and a cutoff-radius
which corresponds to 20 values for µk
, uniformly between 0 and a cutoff-radius  Å. One can attribute
Å. One can attribute  to encode whether the distance between atom i and atom j is close to the value given by µk
. The cutoff function:
to encode whether the distance between atom i and atom j is close to the value given by µk
. The cutoff function:

emphasizes the importance of the local neighborhood by decaying  as a function of rij
and ensures a smooth transition to zero at rc
. For the atoms, we utilize randomly initialized trainable atom type embeddings (figure 2(a),
as a function of rij
and ensures a smooth transition to zero at rc
. For the atoms, we utilize randomly initialized trainable atom type embeddings (figure 2(a),  ). Finally, the bag is represented by the number of remaining atoms (figure 2(a), B).
). Finally, the bag is represented by the number of remaining atoms (figure 2(a), B).

Figure 2. (a) The molecule is represented as a distance matrix D (solid lines between atoms) in a Gaussian basis, embeddings for each atom type  and a bag B. An action constitutes an atom placed in one of the voxels. Each action is represented as a distance to the atoms in the state DQ
(dashed lines, not all distances shown) and a query embedding ZQ
. (b) To predict
and a bag B. An action constitutes an atom placed in one of the voxels. Each action is represented as a distance to the atoms in the state DQ
(dashed lines, not all distances shown) and a query embedding ZQ
. (b) To predict  the network takes as input the state and action representation. The green part of the network operates on the state and the purple section includes the action. The action part is repeated for each voxel in the state resulting in N possible actions. (c) The resulting state after the voxel with the highest Q-value is chosen. If the state is terminal the structure is relaxed using the model potential.
the network takes as input the state and action representation. The green part of the network operates on the state and the purple section includes the action. The action part is repeated for each voxel in the state resulting in N possible actions. (c) The resulting state after the voxel with the highest Q-value is chosen. If the state is terminal the structure is relaxed using the model potential.
Download figure:
Standard image High-resolution imageThe action is represented by distances to atoms already placed (figure 2(a), DQ ) as well as a special query type embedding (figure 2(a), ZQ ). When calculating Q-values the query atom is placed at various voxels allowing for inference of Q-values at possible positions of the next atom to be placed. In this way, all the encoded distances are easily converted to a 3D structure.
The state representation (figure 2(b), green) is updated using SchNet-blocks. In the following we will use superscripts to index the blocks, if multiple blocks follow each other. Fully connected layers are given as Wi
where the full details for each fully connected layer is given in the

where  is elementwise-multiplication and
is elementwise-multiplication and  are the atom embeddings. In the present work we set the dimension of the atom representation to d = 64. As we will process the atom representation further within block l, we use
are the atom embeddings. In the present work we set the dimension of the atom representation to d = 64. As we will process the atom representation further within block l, we use  to denote the intermediate product. The convolution is followed by an atomwise fully connected layer with a skip connection:
to denote the intermediate product. The convolution is followed by an atomwise fully connected layer with a skip connection:

which is passed to the next SchNet block. By chaining several blocks, information from distant atoms are passed to each atom representation. After L = 5 layers, the atomwise representations,  , are used for predicting the energy (E) of the structure as combination of local contributions:
, are used for predicting the energy (E) of the structure as combination of local contributions:

Additionally, we compute a state value (SV):

where  is concatenation. The state value is an indication of whether high or low Q-values are expected from the current state, as will be evident in the next section. Unlike the energy, the state value must consider remaining atoms in the bag when calculating if the current state can progress into a valid molecule.
is concatenation. The state value is an indication of whether high or low Q-values are expected from the current state, as will be evident in the next section. Unlike the energy, the state value must consider remaining atoms in the bag when calculating if the current state can progress into a valid molecule.
Having covered the part of the architecture that deals with the state, we now turn to the action part given in figure 2(b), purple. The query type representation is updated using only a single SchNet-block in order to keep the computational cost low which is desirable since many actions must be queried in every episode. Furthermore, a multi-head self-attention block over all the atom representations follows. In contrast to local models where information from distant atoms must propagate through several blocks, self-attention allows for immediate global information flow. In the case of predicting Q-values for unfinished structures this is especially relevant, as the model must be aware of possible dangling bonds in one end of a molecule while predicting Q-values in the other end. For h = 8 heads, we calculate a query, key and value as:

where the superscript index the head and  are linear projections to a subspace of size
are linear projections to a subspace of size  , where
, where  . From this, a dot-product attention score is calculated:
. From this, a dot-product attention score is calculated:

which indicates the importance of latent representation  to describe
to describe  . From these scores a new latent representation of ZQ
is created as:
. From these scores a new latent representation of ZQ
is created as:

which is finally concatenated and processed by a fully connected layer with a skip connection and layer normalization [45]:

The representation of the query atom is then used for calculating the Q-value:


where  , M is the number of atom types, and
, M is the number of atom types, and  is added to the first M entries of A. Entry i in Q specify the Q-value for atom type i and the
is added to the first M entries of A. Entry i in Q specify the Q-value for atom type i and the  entry allows for low Q-values for all atom types. SV enables the network to lower the Q-value for all possible actions in a given state, instead of independently learning that all actions leads to high-energy structures. By evaluating the Q-value at multiple voxels, the agent follows the greedy policy of picking the action with the highest Q-value, leading to the structure in figure 2(c).
entry allows for low Q-values for all atom types. SV enables the network to lower the Q-value for all possible actions in a given state, instead of independently learning that all actions leads to high-energy structures. By evaluating the Q-value at multiple voxels, the agent follows the greedy policy of picking the action with the highest Q-value, leading to the structure in figure 2(c).
4. Method
The algorithm consists of two phases. In the first phase we use a simple form of imitation learning (IL) called behavioral cloning, where the agent learns to build the structures in the database as well as predicting their energies and forces. This agent serves as a starting point for all agents employed in the next phase.
In the second phase, a RL process is started where stoichiometric-specific agents refines the IL model by constructing new molecules and querying an energy calculator. Based on the received energies and forces, the model updates the Q-values, energies and forces before constructing a new molecule. As the agents receive feedback from the energy calculator the molecules continue to improve.
4.1. Imitation learning phase
To create an initial model the GDB-11 database consisting of molecules containing up to 11 heavy atoms (C, N, O, F) is utilized. Specifically, we use all 1850 structures with 6 heavy atoms for supervised pretraining. Using RDKIT [46] SMILES are transformed into 3D structures and a single point density functional theory (DFT) calculation of the energy and forces is performed using GPAW [47, 48] using a localized atomic basis set [49]. The model is then trained using the following loss function:

where we have:



where the hat denotes values predicted by the network and  ,
,  and
and  are empirically chosen to balance the contributions to the total loss. N is the number of atoms in the structure, with M different atom types.
are empirically chosen to balance the contributions to the total loss. N is the number of atoms in the structure, with M different atom types.
The energy (equation (17)) is trained using a mean squared error (MSE) loss while forces (equation (18)) are updated using a Huber loss given by:

i.e. a squared loss for small differences and a linear loss for large errors. The Huber loss was more stable by suppressing the effect of large force outliers which were occasionally present in the structures built by the model.
Similarly, a cross entropy loss for the Q-values (equation (19)) was found to be more stable than a MSE for the pretraining phase where only Q-values of 0 or 1 are required. For the cross entropy loss  refers to the Q-value for atom type i and we set
refers to the Q-value for atom type i and we set  for the atom type placed in the database and 0 for the other types. Additionally, for each action present in the database we generate five perturbed actions by randomly moving the selected atom and setting
for the atom type placed in the database and 0 for the other types. Additionally, for each action present in the database we generate five perturbed actions by randomly moving the selected atom and setting  for the
for the  th entry in the Q-value prediction, thereby enforcing Q-values for all atom types to be zero for the randomly generated actions. As this generates an imbalance in the training data the valid actions are weighted by a factor of five.
th entry in the Q-value prediction, thereby enforcing Q-values for all atom types to be zero for the randomly generated actions. As this generates an imbalance in the training data the valid actions are weighted by a factor of five.
The model is implemented in Pytorch [50] and trained using the Adam [51] optimizer with an initial learning rate of  and a batch size of 384. We use 90
and a batch size of 384. We use 90 of the structures for training the remaining
of the structures for training the remaining  are used as a validation set for decaying the learning rate by a factor of 2 when the validation error plateaus for 30 epochs. Once the learning rate decayed below 10−6 the validation loss stagnated, and the final model is chosen for employment in the RL phase.
are used as a validation set for decaying the learning rate by a factor of 2 when the validation error plateaus for 30 epochs. Once the learning rate decayed below 10−6 the validation loss stagnated, and the final model is chosen for employment in the RL phase.
4.2. Reinforcement learning phase
In the RL phase we deploy the single IL model for all 135 stoichiometries present in the training set. A cubic cell of dimension [20 Å, 20 Å, 20 Å] and a Q-value grid resolution of 0.2 Å is chosen, with the initial atom restricted to the center of the cell. We use a modified ε-greedy strategy, to trade-off exploration and exploitation. To satisfy two random moves per episode in expectation, we choose a greedy action with probability 1-2/T and a random action with probability 2/T, where T is the number of atoms in the structure. We choose  of random actions uniformly random, while the other
of random actions uniformly random, while the other  are uniformly sampled from the top
are uniformly sampled from the top  of actions. Furthermore, all hydrogen atoms are masked until all other atom types are placed which reduces the action space without excluding any molecules. Due to the IL model an action among the highest Q-values is frequently taken which ensures exploration while simultaneously suppressing the large fraction of actions resulting in completely invalid molecules.
of actions. Furthermore, all hydrogen atoms are masked until all other atom types are placed which reduces the action space without excluding any molecules. Due to the IL model an action among the highest Q-values is frequently taken which ensures exploration while simultaneously suppressing the large fraction of actions resulting in completely invalid molecules.
To limit the action space, we only allow actions which fulfill:

for at least one atom i already present in the structure. Here  ,
,  and
and  is the covalent radius of atom i.
is the covalent radius of atom i.  is the covalent radius of the atom placed in action a and rai
is the distance between the new atom and a present atom i. That is, the new atom must be placed within 0.75–1.25 times the sum of the covalent radii of itself and an already present atom. This restricts the model to building non-fragmented structures for a better comparison with the database.
is the covalent radius of the atom placed in action a and rai
is the distance between the new atom and a present atom i. That is, the new atom must be placed within 0.75–1.25 times the sum of the covalent radii of itself and an already present atom. This restricts the model to building non-fragmented structures for a better comparison with the database.
The agent trained as detailed in the previous section is perfectly capable of building a variety of different molecules given a predetermined stoichiometry. Owing to the stochastic nature of the modified ε-greedy policy employed sequential builds using the same agent results in different structures being built.
We exploit that to construct a first ensemble of structures by tasking the agent with building molecules 200 times. It does so according to the pretrained Q-values and every completed structure is subject to a relaxation in the model energy, equation (8), followed by snapping the molecule back on the grid. If the relaxation results in a fragmentation of the molecule into several pieces, the original structure is used instead. The initial exploration phase promotes exploration of a large part of configuration space to avoid premature convergence to a local minimum. Since the model is pretrained the generated molecules will generally be of high quality.
Once the first 200 builds are completed, the search enters the RL mode. Here the algorithm alternates between generating a structure and updating the model using five mini-batches until a total of 600 additional molecules have been generated, which fine-tunes the agent for each specific stoichiometry. For the Q-values the loss function is changed to MSE, since the target is now given by equation (2), i.e. it is no longer 0 or 1, but depends on the potential energy of the molecule. By further improving the Q-values and force-predictions the search is directed towards low-energy regions of configuration space.
4.3. GDB-11 benchmark
We start by testing the performance of the current framework when it comes to identifying molecules with stoichiometries already present in the database used for the training. The search is conducted as a set of 64 independent restarts following the outlined protocol: using the data-based-trained agent for 200 builds and applying RL for 600 subsequent builds. To facilitate the quantification of the amount of new structures found, we resort to the use of SMILES, as an analytic tool. We stress that SMILES are not used in any way by the agents as this would limit the scope of these. Once the search is finished all structures are post processed by taking five gradient steps using the final model energy landscape. This is a computationally cheap step that removes uncertainties from the original relaxation of the molecules in the less reliable model energy landscape that had been learned up to the point of the RL generating the particular molecule as well as unfortunate grid snapping. These post processed structures are then converted to SMILES and all unique constitutional isomers are found. To this end, we show in figure 3 some of the molecules being built when searching for C4H4O2. As illustrated, we find both new constitutional isomers and often several stereoisomers for these. Looking across these 64 restarts, all of the structures of this composition present in the database (orange arrow) are found again (purple bar). However, also a considerable number of molecules not present in the database are found (green bar).

Figure 3. Subset of discovered constitutional isomers (left) and stereoisomers (right) for C4H4O2. Further shown is the number of all unique constitutional isomers in the database (orange arrow) as well as the molecules produced by ASLA divided into molecules present in the database (purple bar) and novel molecules (green bar).
Download figure:
Standard image High-resolution imageTo investigate whether the produced molecules are of low energy we illustrate the evolution of the 64 agents from episode 400 to 800 in figure 4. The histograms indicate the energy distribution of the structures built in the given episode across the 64 agents. As the search progresses the distribution shifts towards lower energies as the model begins converging, which is further evident by the mean of the distribution (red line) shifting down. Similarly, when we only consider the mean of the lowest energy structure in each run (i.e. not necessarily built in episode 400 or 800), which is depicted using dark red we see a similar shift. In blue, the average energy of the structures in the database, snapped to the grid ASLA operates on, is given. Initially the database is better than the average built of the agent due to imperfect fitting to the database but is overtaken by the agent at episode 800. If one looks only at the best structure in the database (dark blue) the average lowest energy approaches this limit and is marginally beaten by one of the isomers (dark red, dashed). Fully relaxing all unique isomers in DFT indeed reveals that the lowest energy isomer produced by ASLA coincides with the minimum structure in the database.

Figure 4. DFT energy distribution of the molecules. (a) Molecules in the database. Blue line is the average energy and dark blue line is the lowest energy isomer. (b) Orange: structures built in episode 400. The red line indicates the mean energy of the structure and the dark red shows the mean of the lowest energy structure in each run found before episode 400. Finally, the dark red dashed line shows the lowest energy isomer among the 64 parallel runs, found before episode 400. (c) Same as (b), but now at 800 episodes. As the search progresses a shift towards lower energies is observed.
Download figure:
Standard image High-resolution imageTo observe the decision process for the RL algorithm we plot the Q-values for one of the runs in figure 5. First of all, the model has clearly learned a great deal about chemical bonding and thus generally suppresses invalid actions, predicting only high Q-values for sensible bonding sites. The agent is fully aware of possible symmetries during the build. Starting with a single atom in s1, the Q-values have spherical symmetry. For the linear dimer in s2 the symmetry of the Q-values reduces to a ring, and for the Y-shaped C3O backbone in s5, two symmetric 'caps' of high Q values develop. As evident in states s4–s8 where multiple sites have high Q-values, the model decides between different possible isomers when building the molecule. Bond lengths chosen early on e.g. the C–C bond lengths when placing the C atoms from s2 to s5 have an impact on later Q-values. The Q-values encircled by a black ellipse in s8 are thus smaller than the Q-value that are chosen upon building s9. In both cases, aldehyde (HOC-) groups are formed, but at C atoms involved in double or single C–C bonds, respectively, the latter being the energetically preferred. Placing H atoms at both sites would have been possible causing tautomerization involving the methyl group, but again given the initial choices for C–C bond lengths, this is no longer preferable as reflected by the Q-values in s9. Owing to the modified ε-greedy policy, such alternative molecular builds are occasionally made despite the C–C bond lengths, and the subsequent relaxation in the on-the-fly learned model will provide properly adjusted molecules for the RL training.

Figure 5. All Q-values above 0.7 are shown with darker blue corresponding to higher Q-values. For each state we only show the Q-values corresponding to the atom placed in the given step.
Download figure:
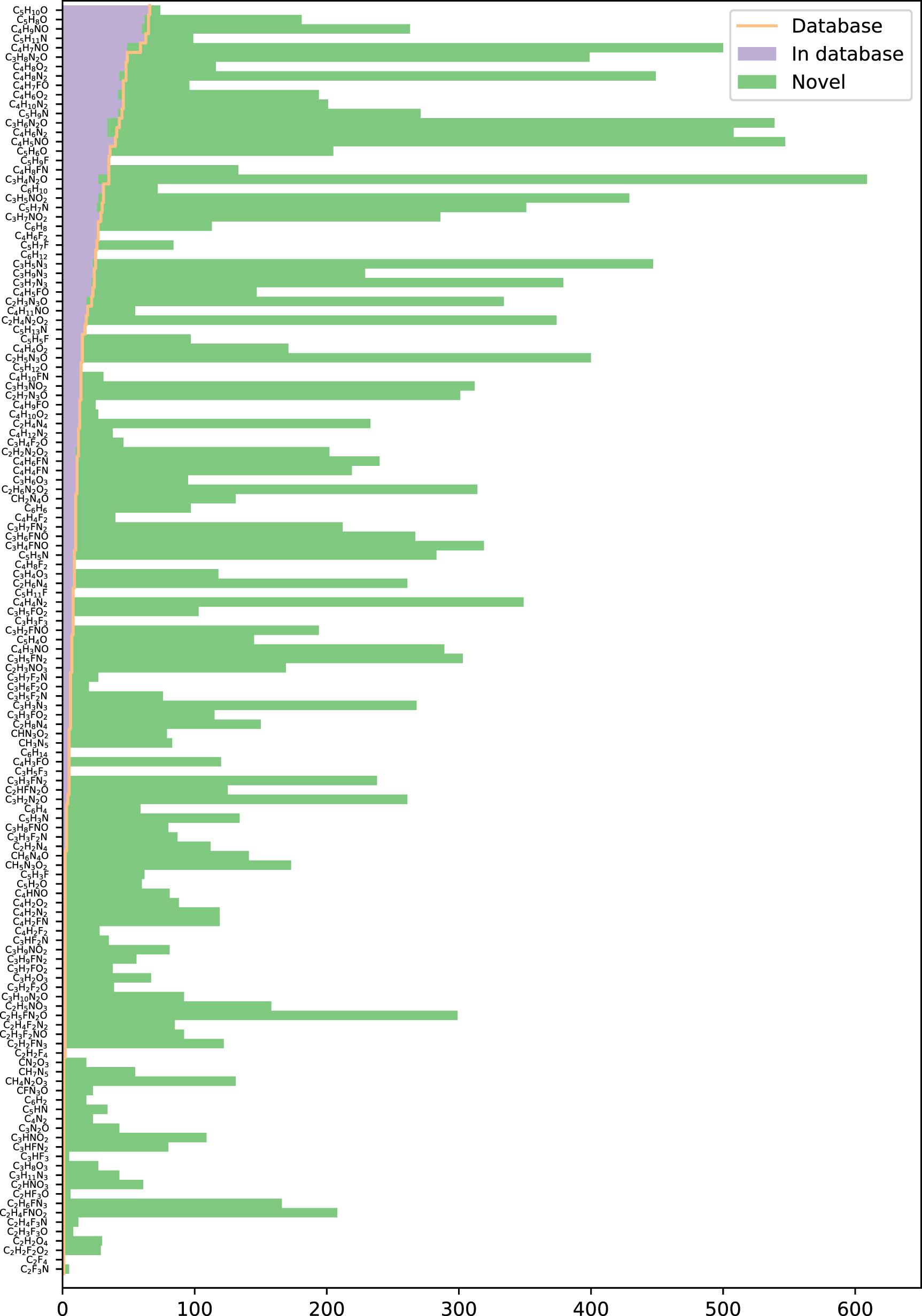
Standard image High-resolution imageIn figure 6 the total number of structures found after 800 episodes for the 15 stoichiometries with most isomers in the database are shown. For a figure involving all 135 stoichiometries, see figure A1 in the

Figure 6. Molecules discovered. Orange line: number of constitutional isomers in the database. Purple bars: molecules found that are also present in the database. Green bars: novel molecules found.
Download figure:
Standard image High-resolution imageIf the RL is neglected and the IL model is used for sampling in all 800 episodes, 1649 of the structures in the database and 15 709 novel molecules are generated for a total of 17 358 unique constitutional isomers. The IL model thus covers a large fraction of the produced structures and turning on RL provides 15.6% more molecules.
To illustrate the difference between the IL model and the RL version, we plot the average atomization energy per atom of the structures generated by ASLA throughout the 800 episodes (figure 7) and compare it to the energies of the structures in the database when placed on the same 0.2 Å resolution grid. For each ASLA curve the average is over the 64 independents run as earlier and now also the 135 stoichiometries. As previously observed, the model trained on the database starts out by producing higher energy structures (red curve) than the average structure in the database (blue curve), due to the imperfect fitting of the database. Similarly, the average of the best structures (dark red) is worse than the lowest energy isomer in the database (dark blue). At around 500 episodes both the average and best structures produced by ASLA marginally outperforms the database. At 800 episodes the best structure is outperformed by 0.04 eV/atom on average. If one only looks at the best structure in each of the 64 runs (dark red, dashed) the database is outperformed by 0.09 eV/atom.

Figure 7. Blue: average energy of structures in the database. Dark blue: average energy of lowest energy molecules in the database. Red: average energy of structures built by ASLA. Dark red: average energy of lowest energy molecules found by ASLA. Dashed dark red: lowest energy isomer among the 64 runs.
Download figure:
Standard image High-resolution imageIn order to check if this energy difference amounts to new low energy structures the specific stereoisomers in the database and the structures generated by ASLA are compared. In that case 1742 of the molecules in the database are found. When counting constitutional isomers as in figure 6, 1769 of the structures in the database were found, which means that in 27 cases the stereoisomer in the database is not found, but the constitutional isomer is. In total, 29 016 stereoisomers not present in the database are found. Each stereoisomer in both the database and generated by ASLA is then fully relaxed using DFT, which shows that for each stoichiometry the lowest energy stereoisomer in the database is found by ASLA for all but one stoichiometry and in 72 of 135 stoichiometries a lower energy stereoisomer is found. It is necessary that both the IL and the RL phases are present, as the IL agent alone will miss some low energy structures whereas as a pure RL procedure will be significantly delayed before discovering the most favourable configurations, see figures A4 and A5 in the
4.4. Exploration beyond the database
Having shown that the ASLA framework is capable of reproducing and expanding molecular structures of compositions already present in the database, we now move to explore its performance when applied outside the realms of the database.
As an example, we investigate C9H8O4, a molecule with twice as many heavy elements as in the training set. As before, 64 independent runs are started.
In figure 8(a) the number of unique constitutional isomers generated as a function of episode number for the IL model (blue) and the model with RL (red) is seen. Unlike for the smaller molecules in IV. C where the IL model was sufficient to cover a large fraction of configuration space there is now a significant difference between the RL and IL agent. The RL model clearly outperforms the IL model in terms of number of molecules generated as the search progresses, showing that the on-the-fly training is able to correct inefficiencies in the initial version. However, the ultimate goal is to generate low energy molecules. Hence, in figure 8(b) the atomization-energy-density of the generated molecules for both the IL model and the RL version is seen. As before all stereoisomers are fully relaxed using DFT. The molecules generated by the RL-enhanced model are significantly more stable than the IL model, with an average energy difference of 0.105 eV/atom or 2.22 eV per structure. Thus, the RL version does not only cover more of configuration space, it is also able to focus on the low energy regions. In figure 9 we investigate a 2D visualization of the generated molecules for both the IL model (squares colored grey to blue) and RL version (circles colored grey to red). The structures are represented using smooth overlap of atomic positions [52] (SOAP) in a 2D space using t-SNE and colored by their energy using two different color schemes to help guide the eye. The 30 lowest energy structures found for both models are framed in black and can be seen figures A2 and A3 in the

Figure 8. Searching for C9H8O4 isomers. (a) Constitutional isomers found across all 64 restarts. As the search progresses, the RL version (red) quickly produces more structures than the IL version. (b) Energy-density of the approximately 5000 and 15 000 unique structures generated by IL and RL, respectively. In addition to producing more constitutional isomers, the RL version (red) generates a larger fraction of low energy molecules.
Download figure:
Standard image High-resolution image
Figure 9. Searching for C9H8O4 isomers. 2D visualization of IL agent (squares) and RL version (circles). All structures are colored by their energy on a scale from grey to blue (IL) or red (RL). The 30 lowest energy structures for both versions of the algorithms are framed in black.
Download figure:
Standard image High-resolution imageFor the IL model we observe fewer of the common aromatic molecules as it has no driving force towards low energy regions of configuration space, as exemplified by the lowest energy isomer found (blue star) being 2 eV higher in energy than the corresponding lowest energy structure in the RL version. The missing focus on low energy parts of configuration space results in the 30 lowest energy structures being scattered across the 2D space, mostly composed of elongated structures (such as the blue square) where subparts, unlike large aromatic rings, are more dominant in the GDB-11 database. Utilizing the feedback from the RL is thus crucial to correct flaws in the IL model as well as expanding beyond knowledge in the database.
5. Conclusion
We have presented a framework for autonomous construction of molecules. The method relies on a preexisting database which via supervised learning provides a base level for a model used for constructing new molecules. Further training of the model is done in a RL setting where feedback is provided via single point energy calculations by a high-level quantum mechanical total energy method. The resulting model is able to reproduce structures in the database as well as producing novel structures. The introduced model is able to operate directly in 3D space allowing for biasing the search towards stable molecules. When applying the IL model in domains far from the training set, the RL procedure corrects initial shortcomings in the model. Further work in this direction could investigate building molecules in specific environments, such as organic light-emitting diode or organic solar panels where properties such as HOMO-LUMO gap could be included in the reward function together with the stability.
Acknowledgments
We acknowledge support from VILLUM FONDEN (Investigator Grant, Project No. 16562). This work has been supported by the Danish National Research Foundation through the Center of Excellence 'InterCat' (Grant Agreement No. DNRF150).
Data availability statement
The data that supports the findings of this study are openly available at the following URL: https://gitlab.au.dk/au143309/data-for-imitation-and-reinforcement-project.
Appendix
Appendix. Network details
In table A1 a detailed overview of the blocks in the network is given. The activation function is a shifted softplus [42] (ssp) given by

Table A1. Dimension of data as it passes through the fully connected layers. The activation function is the shifted softplus indicated by ssp.
| Bag representation | WB | (5, 16, spp, 32) |
| Atomwise layers | Wa | (64, 64, ssp) |
| Distances representation | Wd | (20, 64, ssp) |
| State value | WSA | (96, 32, ssp, 1) |
| Advantage | WA | (96, 32, ssp, 6) |
| Energy | WE | (64, 32, ssp, 16, ssp, 8, ssp, 1) |
| Concatenation layer | Wc | (64, 64) |

Figure A1. Overview of all constitutional isomers built. Orange line indicates number of isomers in the database. Purple are isomers built by ASLA which are also part of the database. Green structures are new isomers.
Download figure:
Standard image High-resolution image
Figure A2. 30 lowest energy structures for the RL run.
Download figure:
Standard image High-resolution image
Figure A3. 30 lowest energy structures for the IL run. Energies are relative to the lowest energy isomer found in the RL version.
Download figure:
Standard image High-resolution image
Figure A4. Histogram of the lowest energy structures for C4N2H6 found for each of 64 independent runs after 3200 episodes using only the imitation learning agent, using reinforcement agents without any imitation learning, and using reinforcement learning agents that start from the imitation learning agent as described in main text. The depicted molecules are exemplify structures at different places in the histogram.
Download figure:
Standard image High-resolution image
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure A5. Histogram of the lowest energy structures for C7H7NO found for each of 64 independent runs after 3200 episodes using only the imitation learning agent, using reinforcement agents without any imitation learning, and using reinforcement learning agents that start from the imitation learning agent as described in main text. The depicted molecules exemplify the structures at different places in the histogram.
Download figure:
Standard image High-resolution image{kind=link}