

Abstract
Somatic mutations in blood indicative of clonal hematopoiesis of indeterminate potential (CHIP) are associated with an increased risk of hematologic malignancy, coronary artery disease, and all-cause mortality. Here we analyze the relation between CHIP status and incident peripheral artery disease (PAD) and atherosclerosis, using whole-exome sequencing and clinical data from the UK Biobank and Mass General Brigham Biobank. CHIP associated with incident PAD and atherosclerotic disease across multiple beds, with increased risk among individuals with CHIP driven by mutation in DNA Damage Repair (DDR) genes such as TP53 and PPM1D. To model the effects of DDR-induced CHIP on atherosclerosis, we used a competitive bone marrow transplantation strategy, and generated atherosclerosis-prone Ldlr−/− chimeric mice carrying 20% p53-deficient hematopoietic cells. The chimeric mice were analyzed 13-weeks post-grafting and showed increased aortic plaque size and accumulation of macrophages within the plaque, driven by increased proliferation of p53-deficient plaque macrophages. In summary, our findings highlight the role of CHIP as a broad driver of atherosclerosis across the entire arterial system beyond the coronary arteries, and provide genetic and experimental support for a direct causal contribution of TP53-mutant CHIP to atherosclerosis
Keywords: somatic, atherosclerosis, clonal hematopoiesis, sequencing
Introduction
Peripheral artery disease (PAD) is a leading cause of cardiovascular morbidity and mortality worldwide1, and age is among its strongest risk factors. PAD associates with an extremely high cardiovascular mortality and unmitigated can progress to limb loss2. The age-related acquisition and expansion of leukemogenic mutations in hematopoietic stem cells has recently been associated with an increased risk of hematologic malignancy, coronary artery disease, and overall mortality3,4. This phenomenon, termed clonal hematopoiesis of indeterminate potential (CHIP), is relatively common in asymptomatic older adults, affecting at least 10% of individuals older than 70 years of age5. CHIP mutations most frequently occur in epigenetic regulators DNMT3A and TET2, in DNA damage repair (DDR) genes PPM1D and TP53, or cell cycle and transcriptional regulator genes JAK2 and ASXL16. CHIP associates with coronary artery disease, stroke and other cardiovascular conditions, but accumulating evidence suggest that the effects of CHIP depend to a great extent on the specific mutated gene and the specific cardiovascular condition 3,7-10. Whether CHIP links with increased risk of atherosclerotic disease in the peripheral arterial bed (PAD) is unknown.
Here, we leveraged 50,122 whole exome sequences from two genetic biobanks (UK Biobank [UKB], Mass General Brigham Biobank [MGBB]) and tested whether CHIP was associated with increased risk of PAD and atherosclerosis across multiple arterial beds, and additionally whether these associations varied by putative CHIP driver gene. Based on these results, we then performed functional analyses in Ldlr-null mice transplanted with 20% Trp53−/− bone marrow (BM) cells, a murine model of atherosclerosis and clonal hematopoiesis driven by TP53 mutations.
Results
Study Cohorts and Risk of Hematologic Malignancy
After excluding individuals with a known history of hematologic malignancy at enrollment, we identified 37,657 unrelated individuals from the UKB and 12,465 individuals from MGBB with whole exome sequencing data available for downstream analysis. Using a previously validated somatic variant detection algorithm11, we identified 2,194 (5.8%) and 657 (5.4%) CHIP carriers in the UKB and MGBB, respectively (Supplementary Table 1). Demographic and clinical characteristics of these individuals, stratified by CHIP status, are depicted in Supplementary Table 2. CHIP carriers tended to be older, male, current or previous smokers, and have a history of coronary artery disease, hypertension, and hyperlipidemia (two-tailed chi-squared and Wilcoxon-rank sum P< 0.05).
We first replicated known CHIP associations11 with white blood cell (Beta 0.09 SD; 95% CI 0.05-0.13; P=1.6x10−5), monocyte (Beta 0.05 SD; 95% CI 0.01-0.09; P=0.009), neutrophil (Beta 0.10 SD; 95% CI 0.06-0.14; P=2.1x10−6), and platelet counts (Beta 0.07 SD; 95% CI 0.03-0.11; P=0.0005) in UKB, with larger CHIP clone size as measured by variant allele fraction (VAF) having stronger effects on blood counts (Extended Data Figure 1). Consistent with the existing literature6,11, CHIP also associated with incident hematologic malignancy (HR 2.20; 95% CI 1.70-2.85; P=1.8x10−9) - specifically for acute myeloid leukemia (HR 8.08; 95% CI 4.36-14.97; P=3.2x10−11), myeloproliferative neoplasms (HR 5.89; 95% CI 3.69-9.89; P=9.7x10−14), and polycythemia vera (HR 12.37; 95% CI 4.85-31.54; P=1.4x10−7). This risk increased with larger VAF (Extended Data Figure 2).
CHIP and Incident PAD Risk
We next tested the association of CHIP status with incident PAD. Using available electronic health record (EHR) data and a previously validated PAD definition12, we identified 338 and 419 incident PAD cases in UKB and MGBB, respectively. CHIP associated with a 58% increased risk of incident PAD in the UKB (HRUKB = 1.58, 95% CI: 1.11-2.25; P=0.01, Figure 1), results that were replicated in MGBB (Overall HR = 1.66, 95% CI: 1.31-2.11; P=2.4x10−5). We then sought to evaluate whether those with larger CHIP clone sizes (i.e., higher VAF) had greater risk for PAD, as larger CHIP clones associate more strongly with adverse clinical outcomes7. We observed a graded relationship between CHIP VAF and PAD, as those with a VAF > 10% had even greater risk for an incident PAD event (Overall HR = 1.97, 95% CI: 1.44-2.71; P=2.3x10−5, Figure 1). Additional sensitivity analyses, including propensity score adjustment and a marginal structural Cox proportional hazards model estimated through stabilized inverse-probability-treatment-weight revealed similar results in the UKB (Extended Data Figure 3). Subsequent analyses showed no significant interaction between CHIP status and either age, sex, or smoking status on incident PAD risk. In sensitivity analyses accounting for a concomitant CAD diagnosis among those with incident PAD, the CHIP-PAD association remained significant (Supplementary Tables 3, 4).
Figure 1. CHIP and incident PAD risk.

a) Association of CHIP and large CHIP (VAF>10%) carrier state with incident PAD events in the UK Biobank (UKB) (N=37,657) and Mass General Brigham Biobank (MGBB) (N=12,465). Results were combined using an inverse-variance weighted fixed effects meta-analysis. Error bars are centered at the HR and show the 95% CI for estimates. b) Cumulative proportion of individuals developing PAD stratified by CHIP VAF clone size category in the UK Biobank (N=37,657). c) Fraction of individuals developing incident PAD by CHIP VAF in the UK Biobank. The dotted vertical line at VAF of 0.10 represents the cut-off for the definition of a large CHIP clone. Error bands reflect the standard error of a generalized binomial additive model with integrated smoothness fit to the data.
CHIP = clonal hematopoiesis of indeterminate potential; VAF = variant allele fraction; PAD = peripheral artery disease
CHIP and Incident Atherosclerosis Across Multiple Vascular Beds
We next assessed whether CHIP was associated with 8 other incident atherosclerotic diseases across multiple vascular beds. Using EHR-based disease definitions13, we tested the association of CHIP with atherosclerotic disease across the mesenteric (acute and chronic), coronary, and cerebral vascular beds, as well as with aneurysmal disease (aortic and any other aneurysm). We observed significant associations for coronary artery disease (HR 1.40, 95% CI: 1.20 to 1.63; P=1.9x10−5), any aortic aneurysm (HR 1.74; 95% CI: 1.21 to 2.51; P=0.0028), other aneurysms (HR 1.70; 95% CI: 1.23 to 2.34; P=0.0013), and mesenteric ischemia (HR 3.22; 95% CI: 2.01 to 5.17; P=1.3x10−6) across both cohorts, with directionally consistent effect estimates observed for all the tested phenotypes (Figure 2a). These associations were consistently stronger for large CHIP clones (Extended Data Figure 4). We then created a composite, incident atherosclerosis outcome combining all nine atherosclerotic phenotypes (“pan-arterial atherosclerosis”, Supplementary Table 5). CHIP associated with this combined incident pan-arterial atherosclerosis endpoint (HR 1.31, 95% CI: 1.14 to 1.49, P=9.7x10−5), again with stronger effects conferred by large CHIP clones (HR 1.45; 95% CI: 1.20 to 1.75; P=0.00013) (Figure 2b,c).
Figure 2. CHIP and incident pan-arterial atherosclerosis risk.

a) Association of CHIP with 9 total incident atherosclerotic diseases separately and combined in a ‘Pan-arterial atherosclerosis’ phenotype in the UKB, MGBB, and meta-analyzed across both studies (“Overall”). Error bars are centered at the HR and show the 95% CI for estimates. b) Cumulative risk of incident atherosclerosis across the composite ‘pan-arterial atherosclerosis’ phenotype stratified by no CHIP, small CHIP (VAF<10%), and large CHIP (VAF≥10%) carrier state in the UK Biobank (N=37,657). c) Association of CHIP VAF with fraction of individuals developing pan-arterial atherosclerosis in the UK Biobank (N=37,657). Error bands reflect the standard error of a generalized binomial additive model with integrated smoothness fit to the data.
CHIP = clonal hematopoiesis of indeterminate potential; VAF = variant allele fraction; PAD = peripheral artery disease
Gene-specific analyses of CHIP with incident atherosclerotic diseases
Next, we sought to understand whether the clonal hematopoiesis putative driver gene differentially affected the risk of acquiring atherosclerosis. Previous work has focused primarily on the epigenetic regulators DNMT3A and TET24,14, and whether DDR CHIP confers an increased risk of atherosclerosis is unknown. We stratified the CHIP-PAD, CHIP-CAD, and CHIP pan-arterial atherosclerosis analyses by putative driver genes and specific mutations - focusing on DNMT3A, TET2, ASXL1, JAK2, the DDR genes PPM1D and TP53, and mutations that specifically disrupt splicing factor genes (LUC7L2, PRPF8, SF3B1, SRSF2, U2AF1, and ZRSR2)15. We observed an association of CHIP with PAD across three of the four common CHIP genes (TET2, ASXL1, and JAK2), with significant heterogeneity of incident PAD effect sizes across the CHIP genes (Pheterogeneity = 0.03) (Figure 3a). This heterogeneity persisted in sensitivity analysis after excluding JAK2 carriers (Pheterogeneity = 0.046). A similar set of associations was observed for CHIP and CAD (Supplementary Table 6). These data also revealed the finding that DDR TP53 and PPM1D CHIP associates with incident PAD (HR 2.72; 95% CI: 1.20 to 1.75; P=0.00013) and incident CAD (HR 2.51; 95% CI: 1.52-4.13; P=0.00032), with a numerically stronger effect on PAD conferred by TP53 mutations (HR 4.98; 95% CI: 1.23-20.09; P=0.024, Figure 3a-c). Similar findings were observed for the incident pan-arterial atherosclerosis outcome when stratifying by putative driver gene (Extended Data Figure 5). Further sensitivity analysis for DDR-CHIP and incident PAD when excluding solid organ malignancy did not significantly change the associations (Pheterogeneity > 0.05).
Figure 3: Gene-specific association of CHIP with incident peripheral artery disease (PAD).

a) CHIP-PAD association analyses stratified by putative CHIP driver gene. Results following meta-analysis across the UKB and MGBB (N=50,122) are shown. Error bars are centered at the HR and show the 95% CI for estimates. b) Gene-specific comparison of HR and 95% CI for hematologic malignancy (x-axis) and PAD (y-axis) in the UKB (N=37,657). Error bars are centered at the HR and show the 95% CI for estimates. c) Association of DDR CHIP (PPM1D or TP53) with incident peripheral artery disease, coronary artery disease, and pan-vascular atherosclerosis. Results across UK Biobank (N=37,657) and MGB Biobank (N=12,465) were combined using an inverse-variance weighted fixed effects meta-analysis. Error bars are centered at the HR and show the 95% CI for estimates. CHIP = clonal hematopoiesis of indeterminate potential; DDR = DNA-damage repair; VAF = variant allele fraction; PAD = peripheral artery disease
Atherosclerosis development in p53−/− CHIP mice
Based on the novel finding of an association between somatic TP53 mutations and atherosclerotic disease, we next characterized further the effects of TP53-mutant CHIP on atherosclerosis through experimental studies in mouse models. The most frequent somatic TP53 variants in CHIP are mainly missense mutations that act in a dominant negative manner and have an effect comparable to that of full p53 deficiency in mouse studies16. Therefore, p53-deficient mice were used as a murine surrogate of human TP53 mutations. To mimic CHIP and test whether the expansion of p53-deficient hematopoietic cells contributes to atherosclerosis, a competitive bone marrow transplantation (BMT) strategy was used to generate atherosclerosis-prone Ldlr−/− chimeric mice carrying 20% Trp53−/− hematopoietic cells (20% KO-BMT mice) (Figure 4a and Supplementary Figure 1). This chimerism is consistent with the VAF of TP53 mutations identified in our human study population (mean VAF=0.13 i.e. ~26% mutant cells if monoallelic mutations; mean VAF=0.23 in those defined as carrying large CHIP clones, Supplementary Table 7). Transplanted mice then consumed a high fat/high cholesterol (HF/HC) diet for 9 weeks to induce atherosclerosis development. To distinguish donor Trp53−/− and Trp53+/+ cells in this experimental setting, Trp53+/+ cells were obtained from mice carrying the CD45.1 variant of the CD45 cell surface marker, whereas Trp53−/− cells were obtained from mice carrying the CD45.2 variant of this protein. Control mice (20% WT-BMT) were transplanted with 20% CD45.2+ Trp53+/+ cells and 80% CD45.1+ Trp53+/+ cells. Flow cytometry analysis of CD45.2+ blood cells established that this BMT strategy led to a modest, but significant expansion of donor Trp53−/− BM-derived cells compared to Trp53+/+ cells in both BM hematopoietic stem/progenitor cells (LSK cells, defined as Lineage- c-Kit+ Sca1+) and circulating white blood cells (Figure 4b,c), consistent with previous studies17-19. Transplanted Trp53−/− BM cells expanded into all blood cell lineages to a similar extent, but this relative expansion did not affect absolute blood cell counts within the timeframe of the experiment (Extended Data Figure 6a,b). Having validated this mouse model of p53-deficient CHIP based on a competitive BMT strategy, we next assessed whether this phenomenon affects the development of atherosclerosis or related metabolic abnormalities. The presence and expansion of Trp53−/− cells led to a significant ~40% increase in plaque size in the aortic root of male Ldlr−/− mice (Figure 4d), without affecting body weight, spleen weight or serum cholesterol levels (Extended Data Figure 6c-e). Similar results were obtained in female mice (Extended Data Figure 6f-k). In contrast to previous findings on DNMT3A/TET2 CHIP14,20-23, the expansion of p53-deficient cells did not affect the expression of the pro-inflammatory cytokines IL-6 and IL-1β or that of the NLRP3 inflammasome in the atherosclerotic aortic wall (Extended Data Figure 7a).
Figure 4. Accelerated atherosclerosis in a murine model of TP53 mutation-driven CHIP.

a) Summary of the competitive BMT approach and the timeline of these studies. 20% KO-BM male mice and 20% WT-BMT controls were fed a high-fat/high-cholesterol (HF/HC) diet for 9 weeks, starting 4 weeks after BMT. b) Percentage of CD45.2+ cells in the BM LSK population (Lin- Sca1+ cKit+ cells) after 9 weeks on HF/HC diet (13 weeks post-BMT), evaluated by flow cytometry (mean±SEM, n=5 20% WT-BMT mice, n=6 20% KO-BMT mice); a two-tailed unpaired t-test was used for statistical analysis. c) Percentage of CD45.2+ cell in the white blood cell population, evaluated by flow cytometry (mean±SEM, n=10 20% WT-BMT mice, n=7 20% KO-BMT mice); a two-way ANOVA with Sidak’s multiple comparison test was used for statistical analysis (** p<0.01; ***p<0.001). Representative CD45.1/CD45.2 dot plots are shown. d) Aortic root plaque size (mean±SEM, n=10 20% WT-BMT mice, n=7 20% KO-BMT mice; a two-tailed unpaired t-test was used for statistical analysis. Representative images of hematoxylin and eosin-stained sections are shown; atherosclerotic plaques are delineated by dashed lines. Scale bars, 100 μm.
Expansion of p53-deficient macrophages in the murine atherosclerotic aorta
Increased atherogenesis in mice carrying Trp53−/− cells was paralleled by a substantial increase in plaque macrophage content, as assessed by immunohistological staining of Mac2 (Figure 5a), suggesting a contribution of increased arterial macrophage burden to accelerated atherosclerosis in conditions of p53-deficient CHIP. No significant changes were observed in other plaque components, such as collagen content, vascular smooth muscle cell content, necrotic core extension or lipid content. (Figure 5a and Extended Data Figure 7b). Flow cytometry analysis of matched samples from blood and digested atherosclerotic aortae from 20% KO-BMT mice revealed substantially higher chimerism in aortic macrophages (~76% Trp53−/−) than in blood classical monocytes (~48%), the main source of plaque macrophages (Figure 5b). Immunofluorescent staining of aortic root sections confirmed that plaque macrophages are predominantly Trp53−/− in 20% KO-BMT mice (Figure 5c). These findings suggest that p53-deficient macrophages have a selective advantage to expand within the atherosclerotic plaque.
Figure 5. Expansion of p53-deficient macrophages within atherosclerotic plaques.

a) Histological analysis of plaque composition in 20% KO-BMT female mice and 20% WT-BMT controls, quantified as absolute intimal content of macrophages (Mac2 antigen immunostaining), vascular smooth muscle cells (smooth muscle α-actin, SMA immunostaining), collagen (Masson’s trichrome staining) and necrotic core (collagen-free acellular regions); representative images are shown (mean±SEM, n=10 20% WT-BMT mice, n=8 20% KO-BMT mice). Two-tailed unpaired t-tests were used for statistical analysis. b) Percentage of CD45.2+ cells within the aortic macrophage population (CD3−, Ly6G−, CD11b+, F4/80High) and blood classical monocytes (CD3−, CD115High, Ly6G−, CD43Low, Ly6CHigh) of 20% KO-BMT mice and controls, evaluated by flow cytometry (mean±SEM, n=6 independent mice per BM genotype in classical monocyte analyses, n=3 pools of 2 aortic arches collected from independent mice in aortic macrophage analyses). A two-way ANOVA with Tukey’s multiple comparison test was used for statistical analysis Representative CD45.1/CD45.2 plots of aortic macrophages are shown. c) Immunofluorescent staining and confocal microscopy imaging of CD45.2+ cells (green) and CD68+ macrophages (red) in atherosclerotic plaques of 20% KO-BMT mice and 20% WT-BMT controls. DAPI-stained nuclei are shown in blue. Representative images are shown (sections from n=14 20% WT-BMT mice and n=12 20% KO-BMT mice from 2 independent experiments were examined). Yellow triangles indicate examples of CD45.2/CD68-doble positive cells. Atherosclerotic plaques are delineated by dashed lines. Scale bars, 50 μm.
Increased proliferation of murine p53-deficient macrophages
Atherosclerotic plaques in mice carrying p53-deficient hematopoietic cells exhibited a significant increase in the proportion of cells positive for Ki-67, a marker of proliferating cells (Extended Data Figure 8a). Flow cytometry analysis indicated that this is at least in part driven by increased proliferation of p53-deficient macrophages, as there was a 2-fold increase in the frequency of Ki-67+ cells within the CD45.2+ Trp53−/− aortic macrophage population compared to CD45.2+ Trp53+/+ macrophages (Figure 6a). Consistent with this in vivo observation, cultured Trp53−/− macrophages exhibited accelerated mitotic cell cycle progression, with a >2-fold increase in the percentage of DNA-replicating (S-phase) cells upon stimulation with macrophage colony stimulating factor (MCSF), a major determinant of plaque macrophage proliferation24 (Figure 6b). Furthermore, Trp53−/− macrophages showed and increased percentage of S-phase cells when proliferating asynchronously and higher rates of BrdU incorporation when stimulated with MCSF (Extended Data Figure 8b,c). p53 was expressed in quiescent macrophages at the transcript and protein level and further induced after MCSF stimulation (Figure 6c,d), suggesting a central role of p53 in the normal regulation of macrophage cell cycle progression. Consistent with this possibility, transcriptomic profiling by RNA-seq revealed a widespread alteration of gene expression in MCSF-stimulated Trp53−/− macrophages compared to WT controls (Extended Data Figure 9). Functional annotation of differentially expressed genes showed that categories related to quantity of myeloid cells, cell cycle progression and immune cell proliferation were among the most upregulated in Trp53−/− macrophages (Extended Data Figure 9). qPCR analysis at different timepoints of MCSF stimulation further confirmed that p53-deficient-macrophages exhibit major changes in the expression of pivotal regulators of cell cycle entry and progression, such as Cdkn1a/p21Cip1 and Cyclin B1 (Figure 6e). RNA-seq of cultured p53-deficient macrophages also revealed a significantly downregulated expression of several genes related to cell death and apoptosis (Extended Data Figure 9). However, apoptosis rates were comparable in p53-deficient and WT macrophages within atherosclerotic plaques (Supplementary Figure 2), suggesting that differences in apoptosis are not a relevant contributor to increased macrophage burden and atherosclerotic plaque size in mice exhibiting p53-deficient CHIP. Accordingly, we found that p53 deficiency does not affect oxysterol-induced apoptosis in cultured macrophages (Supplementary Figure 2). Similarly, neither the uptake of modified LDL, the uptake of apoptotic cells, or the expression of central mediators of these cellular processes were affected by p53 deficiency (Supplementary Figure 3). p53-deficient neutrophils isolated from 20%-KO-BMT mice exhibited an expression of phenotypic markers and a production of reactive oxygen species comparable to those of WT neutrophils (Supplementary Figure 4). Neutrophil extracellular traps were not detected in murine atherosclerotic plaques in the experimental conditions used in this study (Supplementary Figure 5).
Figure 6. Increased proliferation of p53-deficient macrophages.

a) Percentage of Ki-67+ proliferating cells within the aortic CD45.2+ macrophage population of 20% KO-BMT mice and controls evaluated by flow cytometry (mean±SEM, n=3 pools of 2 murine aortic arches per BM genotype) evaluated by flow cytometry; a two-tailed unpaired t-test was used for statistical analysis. Representative plots are shown. b) % of S-phase cells in cultures of Trp53−/− and +/+ murine BM-derived macrophages, evaluated by propidium iodide staining of cellular DNA content and flow cytometry. Treatment with MCSF was used to induce cell cycle entry and progression in quiescent G0-synchronized macrophages (mean±SEM, n=10 Trp53+/+ mice, n=9 Trp53−/− mice). A two-way repeated measures ANOVA with Sidak’s multiple comparisons test was used for statistical analysis. c, d) qPCR (c, n=7 mice) and Western Blot (d, n=5 mice) analyses of Trp53 expression in cultured primary macrophages after MCSF mitogenic stimulation. A representative blot is shown. (mean±SEM, *p<0.05; ****p<0.0001 vs baseline); a one-way repeated measures ANOVA with Tukey’s multiple comparison test was used for statistical analysis. e) Expression of cell cycle regulators Cdkn1a/p21Cip1 and Ccnb1/Cyclin B1 in cultured Trp53−/− and +/+ macrophages proliferating asynchronously (Async, mean±SEM, n=10 mice per genotype) or after MCSF stimulation (mean±SEM, n=7 Trp53+/+ mice, n=6 Trp53−/− mice). A two-tailed unpaired t-test (with Welch’s correction for Ccnb1 analysis) and a two-way repeated measures ANOVA with Sidak’s multiple comparison test, respectively, were used for statistical analysis (**p<0.01; ***p<0.001; ****p<0.0001).
Discussion
This study combined exome sequencing data across two biobanks to detect somatic mutations in over 50,000 individuals and observed that the presence of CHIP was significantly associated with an increased risk of developing PAD and atherosclerosis across multiple arterial beds. Increased risk was differentially observed across CHIP driver genes with evidence of a graded relationship with CHIP VAF, with large CHIP clones conferring greater risk of disease. The scale of this study allowed a high-power analysis of gene-specific associations for a variety of CHIP genes, which provided evidence of an association between TP53-mutant CHIP and the risk of incident atherosclerotic disease, including both CAD and PAD. Lastly, experimental studies in mice demonstrated that p53-deficient CHIP results in increased size of aortic atherosclerotic plaques, accompanied by expansion of plaque macrophages, supporting a direct contribution of p53-mutant hematopoietic cells to accelerated atherogenesis.
These findings permit several conclusions. First, in humans, CHIP appears to promote atherosclerosis across the entire arterial system, but its effects vary by putative CHIP driver gene. Previous work linked CHIP with increased risk of coronary artery disease and early-onset MI3,4. We build on these findings by demonstrating that CHIP is also associated with PAD and a composite pan-arterial atherosclerosis outcome reflective of an increased burden of atherosclerosis throughout the human arterial system. Our results demonstrate significant evidence of heterogeneity in CHIP effects based on putative driver gene, which is particularly evident when evaluating the effects of DNMT3A mutations. Jaiswal et al demonstrated an increased risk of coronary artery disease among individuals with DNMT3A-mutant CHIP4, but we did not observe a significant effect on the risk of CAD or PAD, despite the fact that DNMT3A mutations were the most frequent mutations in our study population. As the field of CHIP continues to evolve, shifting the focus from CHIP as a broad entity to deciphering the specific effects of individual CHIP driver genes will be critical.
Second, DDR gene CHIP appears to confer an increased risk of atherosclerotic cardiovascular disease. In previous work, Jaiswal et al demonstrated an increased risk of coronary artery disease among individuals with CHIP through DNMT3A, TET2, ASXL1, and JAK2 somatic driver mutations4. However, it remained unknown whether individuals who exhibit CHIP driven by mutations in DDR genes have a heightened risk of developing atherosclerotic disease. This gap in knowledge is relevant, as DDR CHIP can be detected in a substantial number of cancer-free individuals and is particularly frequent in cancer patients and survivors treated with cytotoxic therapies, a population at high CVD risk25. In this context, the current study demonstrates that CHIP related to DDR-genes (TP53, PPM1D) confers an increased risk of developing atherosclerosis, both in coronary and peripheral arteries. These findings lend human genetic support to the concept that post-cytotoxic therapy patients may benefit from surveillance for atherosclerotic conditions in addition to therapy-related myeloid neoplasms. Future studies will be required to assess whether DDR-CHIP contributes to the increased CVD risk in cancer patients and survivors.
Third, CHIP related to TP53 mutations appears to drive atherosclerosis risk via expansion of p53-deficient macrophages within atherosclerotic plaques. Previous experimental studies assessed the role of p53 in atherogenesis using mice engineered to exhibit gain or loss of function of p53 in the whole body or in specific cell types.26-32 Overall, these studies suggested that p53 exerts atheroprotective actions28-30, although some null findings have also been reported26,31. Here we demonstrate that carrying a fraction of p53-deficient blood cells is sufficient to accelerate atherosclerosis development. Although the contribution of additional mechanisms cannot be ruled out, this accelerated atherosclerosis seems mainly related to an increase in macrophage burden in the atherosclerotic plaque, as p53-deficient macrophage have a selective advantage to expand within the arterial wall. Mechanistically, this expansion of p53-deficient macrophages seems mainly related to increased proliferation, a major driver of macrophage burden in atherosclerotic plaques33. Although p53 expression is typically induced by DNA damage or other kinds of cellular stress, we found that it is expressed in resting conditions in cultured macrophages and further induced by mitogenic stimulation, suggesting a physiological role in the regulation of macrophage cell cycle progression in the absence of stressors. Accordingly, p53-mutant macrophages exhibited accelerated cell cycle kinetics. Overall, these experimental findings provide support to the notion that TP53 CHIP mutations contribute directly to accelerated atherosclerosis, and highlight major differences in the mechanisms underlying accelerated atherosclerosis in CHIP driven by mutations in TP53 or epigenetic regulators. In contrast to previous reports related to CHIP driven by mutations in the epigenetic regulatory genes DNMT3A, TET2 and ASXL114,20-23,34, we did not observe a significant effect of p53 deficiency on the expression of the pro-inflammatory cytokines IL-1β and IL-6. These results are consistent with previous human data showing that circulating levels of pro-inflammatory cytokines are significantly increased in carriers of CHIP driven by mutations in these epigenetic regulators, but not in carriers of mutations in TP53 or PPM1D11. These mechanistic differences between TP53-CHIP and DNMT3A/TET2/ASXL1-CHIP require consideration when designing preventive care strategies targeting the effects of CHIP on atherosclerosis. Thus, while the pathogenic effects of TET2-CHIP may be prevented by targeting IL-1β-driven inflammation14, accelerated atherosclerosis associated with TP53 mutations may be better tackled by other strategies. Additional experimental and clinical studies should evaluate these opportunities for personalized medicine in the context of CHIP.
Lastly, a recent publication has generated a novel hypothesis regarding the relationship between CHIP and atherosclerosis35. In their analysis, Heyde et al. suggest that CHIP may be a symptom of the atherosclerosis trait complex (the interplay of chronic inflammation, hyperlipidemia, and arterial plaque), rather than a causal risk factor, based on murine and human observations that atherosclerosis may accelerate HSC proliferation and mutant clone evolution. While their study engenders an intriguing hypothesis, some of the observations in our current study contradict this interpretation. Heyde et al. state that the uniform hazard ratios previously observed for the CHIP-CAD association provide supportive evidence for the notion that atherosclerotic disease precedes the development of clonal hematopoiesis. However, in our study we identify significant heterogeneity of incident PAD and CAD effect sizes when stratifying by CHIP genes, as well as a dose-response relationship between CHIP clone size (or VAF) and incidence of atherosclerosis, suggesting that CHIP genes vary in their effect on incident PAD risk, and that expanded CHIP variants have a stronger influence on future atherosclerotic risk. Thus, given the 1) association of CHIP with incident atherosclerotic disease, 2) dose-response relationship with CHIP clone size and heterogeneity of effect size by CHIP gene, and 3) that mutations in TP53, TET2, and JAK236 appear to drive atherosclerosis through disparate mechanisms, clonal hematopoiesis acting as a causal risk factor, at least when driven by certain mutations, appears to be a more parsimonious explanation to explain the association between CHIP and the risk of atherosclerotic disease
Several limitations merit mention. First, our atherosclerotic disease phenotypes are based on EHR data and may result in misclassification of case status. Such misclassification would likely reduce statistical power for discovery and on average bias results toward the null. Second, selection bias from differential loss-of-follow up, volunteer bias, and missingness in covariates may be present given the nature of the genetic biobanks used in this study. Third, while we maximized the number of participants in our analysis of CHIP and additional atherosclerotic diseases, it may still have been underpowered for certain phenotypes. Fourth, while TP53 somatic mutations most commonly occur in post-cytotoxic chemotherapy patients, we did not specifically examine this subset of individuals given their small sample size and future studies will be needed to validate these findings in this population. Lastly, our mouse model of CHIP was based on p53-deficient homozygous cells, whereas most cancer-free individuals with TP53-mutant CHIP can be expected to carry monoallelic mutations. However, the most frequent TP53 mutations linked to CHIP act in a dominant negative manner and, even when monoallelic, lead to phenotypes similar to full biallelic TP53 loss of function, as shown previously16. Thus, Trp53−/− cells provide and overall appropriate model of these CHIP-related mutations. Additional studies will be required to assess the effects of specific TP53 mutations on atherosclerosis.
In conclusion, CHIP, including TP53-CHIP, is associated with incident atherosclerosis disease in humans, and experimental mouse studies suggest a direct contribution of somatic TP53 mutations to atherosclerosis development through an expansion of plaque macrophages. These observations expand our knowledge of the pathophysiological relevance of CHIP in atherosclerosis and highlight the value of single gene analyses that shed light into the effects of mutations in specific CHIP driver genes.
Methods
Cohorts and exclusion criteria
The UKB is a population-based cohort of approximately 500,000 participants recruited from 2006-2010 with existing genomic and longitudinal phenotypic data and median 10-year follow-up37. Baseline assessments were conducted at 22 assessment centres across the UK with sample collections including blood-derived DNA. Of ~49,960 individuals with WES data available, we analysed 37,657 participants consenting to genetic analyses after our exclusion criteria. Use of the data was approved by the Massachusetts General Hospital Institutional Review Board (protocol 2013P001840) and facilitated through UK Biobank Application 7089.
The MGBB contains genotypic and clinical data from >105,000 patients who consented to broad-based research across 7 regional hospitals and median 3-year follow-up38. Baseline phenotypes were ascertained from the electronic medical record and surveys. We analysed 12,465 individuals consenting to genetic analysis after our exclusion criteria. Use of the data was approved by the Massachusetts General Hospital Institutional Review Board (protocol 2020P000904).
Across both cohorts, we excluded individuals with prevalent hematologic cancer, individuals without genotypic-phenotypic sex concordance, and one of each pair of 1st or 2nd degree relatives at random. For the UKB, samples were further restricted to individuals with Townsend deprivation index, a marker of socioeconomic status, available for analysis. Follow-up time was defined as time from enrolment to disease diagnosis for cases, or to censorship or death for controls.
Whole exome sequencing and CHIP calling
UKB WES were generated from whole blood-derived DNA at the Regeneron Sequencing Center39. MGBB WES were generated using whole blood-derived DNA using Illumina sequencing (mean coverage 55x). Somatic CHIP variants were detected with GATK MuTect2 (v1) software with parameters as previously described7,11. Briefly, aligned short-read exome sequences were analyzed using the GATK MuTect2 software to detect putative somatic genetic variants, which were then filtered to exclude common sequencing artifacts (including minimum variant read counts of 3 reads, required evidence of a variant on both forward and reverse reads, exclusion of frameshift mutations within homo-polymer repeat regions). Common germline variants and sequencing artifacts were excluded as before. Samples were annotated with the presence of any CHIP if MuTect2 identified one or more of a pre-specified list of pathogenic somatic variants reported in the literature and/or the Catalog of Somatic Mutations in Cancer (COSMIC, http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/ ), following manual curation, as previously described21,40. Additionally, samples were annotated with the presence of Large CHIP (variant allele frequency >10%), as larger CHIP clones have previously been more strongly associated with adverse clinical outcomes7.
Phenotype definitions
Across both UKB and MGBB, PAD was defined by grouping together ICD-10 and ICD-9 billing codes for aortic atherosclerosis (I70), peripheral vascular disease (I73.8, I73.9), and operative procedures including amputation of leg (X09.3-5), bypass of artery of leg (L21.6, L51.3, L51.6, L51.8, L59.1-8), endarterectomy or angioplasty of leg artery (L52.1-2, L54.1,4,8, L60.1-2, L63.1,5), and other transluminal operations on leg arteries or peripheral stent placement (L63.9,L66.7), as previously described12. Additionally, in the UKB self-reported peripheral vascular disease, leg claudication/intermittent claudication, arterial embolism, femoral-popliteal leg artery bypass, leg artery angioplasty +/− stent, or amputation of leg were also included (Data fields 20002, 20004) as performed previously12. Coronary artery disease and cerebral atherosclerosis phenotype definitions for each cohort are detailed in Supplementary Tables 8 and 9. Other atherosclerotic conditions were defined using the Phecode Map 1.241 ICD-9 (https://phewascatalog.org/phecodes) and ICD-10 (https://phewascatalog.org/phecodes_icd10) phenotype groupings for “abdominal aortic aneurysm”, “aortic aneurysm”, “other aneurysm”, “chronic vascular insufficiency of intestine”/“acute vascular insufficiency of intestine” (combined into a mesenteric ischemia phenotype), “atherosclerosis of renal artery”. The composite atherosclerosis phenotype was created by combining all analyzed atherosclerosis phenotypes (including PAD, coronary artery disease, cerebral atherosclerosis, abdominal aortic aneurysm, aortic aneurysm, other aneurysm, chronic vascular insufficiency of intestine, acute vascular insufficiency of intestine, and atherosclerosis of renal artery) into one phenotype, whereby the first instance across all of these phenotypes was used to determine time of first diagnosed atherosclerotic disease for survival analysis. Other phenotypic covariates (never/prior/current smoking status, hypertension, hyperlipidemia, principal components of ancestry, etc.) were used as previously defined42.
Association Analysis
In the UKB and MGBB a traditional cox-proportional hazards model was utilized using the Survival package in R-3.5 adjusting for age, age2, sex, smoking status, normalized Townsend deprivation index as a marker of socioeconomic status (only available in UKB), and the first ten principal components of genetic ancestry. Demographic and clinical characteristics found to differ between individuals with and without CHIP were tested using chi-squared (categorical) and Wilcoxon-rank sum (continuous) tests with a two-tailed P< 0.05 determining significance. Sensitivity analysis including other covariates (normalized BMI, prevalent hypertension, prevalent Type 2 diabetes, and prevalent hyperlipidemia) was not found to significantly change associations with PAD in the UKB (Extended Data Figure 10). Additional sensitivity analyses were utilized in the UKB including propensity score adjustment, as well as a marginal structural cox proportional hazards model estimated through stabilized inverse-probability-treatment-weight (IPTW)43 to estimate the total causal effect of CHIP on PAD. Further details of propensity score methods and stabilized IPTW analysis are described below. For our primary outcome PAD, results were combined across the UKB and MGBB using an inverse-variance weighted fixed effects meta-analysis, a two tailed association P < 0.05 determined statistical significance. In our secondary analysis of CHIP with 10 additional, incident atherosclerotic diseases, a two-tailed Bonferroni p-value threshold of P<0.05/10=0.005 was used to declare statistical significance.
Epidemiologic causal inference methods used to assess the CHIP-PAD association
The propensity score is the probability of treatment assignment (i.e. clonal hematopoiesis of indeterminate potential [CHIP] or large CHIP) conditional on the minimally sufficient set of confounders. Using a propensity score as a covariate allows observational studies to mimic some of the characteristics of a randomized controlled trial in the sense that conditional on the propensity score, the distribution of covariates will be similar between the CHIP and the non-CHIP carriers. In our scenario, the propensity score will help assess the ‘dimensionality problem’ in that it will reduce the number of covariates from 15 covariates in the minimally sufficient set to 1. Here, the propensity score is calculated using multivariable logistic regression where the outcome was CHIP and covariates consisted of the 15 covariates in the minimally sufficient set (age, age2 – included to fully account for age given significant associations in multivariate models, sex, smoking status, and the first ten principal components of genetic ancestry). The propensity score was then predicted for all samples in the UK Biobank for CHIP and, separately, for large CHIP.
Stabilized Inverse probability of treatment weighting (IPTW) uses weights based on the propensity score to create a pseudo-population in which the distribution of measured covariates is independent of CHIP status, i.e. removing the arrow from the confounders to the exposure. The conditional exchangeability assumption made in IPTW is that if the set of covariates are sufficient to block all backdoor paths from CHIP to peripheral artery disease (PAD), then all confounding is eliminated in the pseudo-population and thus the association between CHIP and PAD in the synthetic population is an estimate of the causal effect. To estimate the IPTW stabilized weight (SW) for each individual in the UKB, we utilized the following equation for CHIP carriers and controls, where PS reflects the propensity score for that individual:
Mice
C57Bl/6J Trp53−/− mice were obtained from the Jackson Laboratory. Ldlr−/− mice carrying the CD45.1 isoform of the CD45 hematopoietic antigen were generated by crossing Ldlr−/− mice from the Jackson Laboratory and B6.SJL-PtprcaPepcb/BoyCrl mice from Charles River Laboratories. Animal experiments followed protocols approved by the Institutional Ethics Committee at the Centro Nacional de Investigaciones Cardiovasculares and conformed to EU Directive 86/609/EEC and Recommendation 2007/526/EC regarding the protection of animals used for experimental and other scientific purposes, enforced in Spanish law under Real Decreto 1201/2005. All mice were maintained on a 12-h light/dark schedule in a specific pathogen-free animal facility in individually ventilated cages, and were given food and water ad libitum. Ambient temperature in the animal facility was 20-24°C, relative humidity was 45-65%.
Competitive bone marrow transplantation and atherosclerosis induction in mice
8-12 weeks-old CD45.1+ Ldlr−/− recipients were transplanted with suspensions of BM cells containing 20% CD45.2+ Trp53−/− cells and 80% CD45.1+ Trp53+/+ cells (20% KO-BMT mice) or 20 % CD45.2+ Trp53+/+ cells and 80% CD45.1+ Trp53+/+ cells (20% WT-BMT mice) similar to previous studies14 (Supplementary Figure 1). BM cells were isolated from femurs and tibias of 6-12 weeks-old donor mice after euthanasia. Recipient Ldlr−/− mice were exposed to two doses of 450 rad three hours apart. After the second irradiation, each recipient mouse was injected with 107 BM cells i.v. Water was supplemented with antibiotics for 7 days before transplant and for 14 days post-transplant. Mice that did not recover full pre-irradiation body weight 28 days after transplant were excluded from further analysis. Starting four weeks after BMT, an interval sufficient for BM reconstitution in our experimental conditions, mice were fed a HF/HC Western diet (Harlan-Teklad, TD.88137, Adjusted Calories Diet; 42% from fat, 0.2% cholesterol) to promote hypercholesterolemia and the development of atherosclerosis. Mice were maintained on HF/HC diet for 9 weeks unless otherwise stated.
Analysis of aortic atherosclerosis burden in mice
Mice were euthanized and plasma was collected in EDTA-coated tubes for cholesterol level quantification by an enzymatic assay (Cholesterol E, WAKO Diagnostics). Aortas were removed after in situ perfusion with phosphate-buffered saline (PBS) injected through the left ventricle of the heart. Tissue fixation was achieved by immersion in 4% paraformaldehyde in PBS overnight at 4°C. Aortic tissue was then embedded in paraffin to obtain histological sections from the aortic root as determined by the location of the aortic valve leaflets. Alternatively, unfixed aortic root specimens were embedded in Tissue-Tek O.C.T. Compound. An operator who was blinded to genotype quantified plaque size in aortic root sections by computer-assisted morphometric analysis of microscopy images. For each mouse, atherosclerotic plaque size in aortic root cross-sections was calculated as the average of 5 independent sections separated by ~16μm. Atherosclerotic plaque composition was examined by immunohistochemical techniques. Vascular smooth muscle cells were identified with an alkaline phosphatase-conjugated mouse anti-smooth muscle α-actin (SMA) monoclonal antibody (clone 1A4, Sigma) and Vector Red Alkaline Phosphatase Substrate (Vector Laboratories). Macrophages were detected with a rabbit anti-Mac2 monoclonal antibody (Cedarlane Labs), a biotin-conjugated goat anti-rat secondary antibody, streptavidin-HRP, and DAB substrate (all from Vector Laboratories), with hematoxylin counterstaining. Ki-67 was detected with a rabbit anti-Ki-67 antibody (clone SP6, Abcam), a biotin-conjugated horse anti-rabbit secondary antibody and streptavidin HRP. Collagen and lipid content were determined by a modified Masson’s trichrome staining and Oil Red O (ORO) staining, respectively. Microscopy images were analyzed using Fiji ImageJ software using the Color Deconvolution plugin. Plaque cell apoptosis was quantified by TUNEL analysis (In situ Cell Death Detection Kit, Fluorescein, Roche) according to manufacturer instructions. Prior to apoptotic cell staining, sections where blocked with 3% of goat serum in PBS and co-stained with Alexa Fluor 647-conjugated anti-CD45.2 antibody (BioLegend), anti-CD68 primary antibody (BioRad) and Alexa Fluor 568-conjugated goat anti-rat IgG secondary antibody (ThermoFisher). Nuclei were counterstained with DAPI. Images were acquired on a LEICA SP8 Lightning-Navigator confocal microscope with LAS X 3.1.1. 15751 software, using a z-stack of 5 μm-thickness frames. Results are shown as the percentage of TUNEL positive cells within the total plaque cell population, the plaque macrophage population or the CD45.2+ macrophage population in the aortic root. A list of antibodies and additional information, including antibody dilutions, can be found in Supplementary Table 10.
Flow cytometry analyses of blood and tissue samples.
Peripheral blood was obtained from the facial vein and collected into EDTA-coated tubes. BM cells were flushed out of two femurs and two tibias per mouse. Aortic arches were digested for 45 minutes at 37°C in RPMI containing 10% FBS and 0.25 mg/ml Liberase TM (Roche Life Science). Red blood cells were lysed in all samples by treatment with 1X Red Blood Cell Lysis Buffer (eBioscience ThermoFisher) for 5 minutes on ice. BM lineage-negative cells were defined as negative for CD11b, Gr-1, Ter119, B220, CD3e and CD127. BM LSK were defined as Lineage−, c-Kit+, Sca1+. Blood classical monocytes were identified as CD45+, CD115High, CD43Low, Ly6CHigh; patrolling monocytes, as CD45+, CD115High, CD43High, Ly6CLow; neutrophils, as CD45+, CD115Int, Ly6G+; T lymphocytes, as CD45+, CD115−, B220−, CD3+; B lymphocytes, as CD45+, CD115−, CD3−, B220+; and aortic macrophages, as CD45+, CD3−, Ly6G−, CD11b+, F4/80High. Gating strategies are summarized in Supplementary Figure 6. Samples were stained with combinations of biotinylated and/or fluorescently labeled antibodies in PBS with 1% FBS for 30 min on ice. The following fluorescent antibodies were used for staining and flow cytometry analysis (additional information, including antibody dilutions, can be found in Supplementary Table 10): eFluor450-conjugated anti-CD45.2, FITC-conjugated anti-CD45.2, Pe-Cy7-conjugated anti-CD45.1, FITC-conjugated anti-CD11b, eFluor450-conjugated anti-CD11b, PE-conjugated anti-CD115, PE-eFluor610-conjugated anti-CD3, FITC-conjugated anti-CD4 (from eBioscience); APC-Cy7-conjugated anti- B220, PerCP-Cy5.5-conjugated anti-Ly6G, BV510-conjugated anti-Ly6G, BV711-conjugated anti-CD43, PE-Cy7-conjugated anti-c-Kit, AlexaFluor647-conjugated anti-Sca-1 (from BD Biosciences); PE-conjugated anti-F4/80 (from R&D Systems); PerCP-Cy5.5-conjugated anti-CD45.1, BV510-conjugated anti-CD8a, PerCP-Cy5.5-conjugated anti-mouse Ki-67 (from BioLegend). Fixation/permeabilization for Ki-67 intracellular staining was achieved using commercially available kits following manufacturer’s instructions (Foxp3 Transcription Factor Staining Buffer Set, eBioscience ThermoFisher). Dead cells were excluded from analysis by DAPI staining in unfixed samples and by LIVE/DEAD Fixable Near-IR staining (ThermoFisher Scientific) in fixed samples. BD LSRFortessa and BD FACSymphony Cytometers were used for data acquisition with BD FACSDiva software (BD Biosciences). Data were analyzed with FlowJo Software (Tree Star).
Murine bone marrow-derived macrophage culture and cell cycle analysis
BM-derived macrophages (BMDM) were obtained by differentiating femoral BM cell for 7 days in RPMI Medium supplemented with antibiotics, 10% fetal bovine serum and 100 ng/ml MCSF. Cell-cycle dynamics were analyzed both in asynchronously proliferating macrophages and after synchronization in G0 phase by MCSF deprivation. MCSF concentration in cell culture medium was decreased to 5 ng/ml for 48h to induce progressive synchronization of macrophages in G0 phase (i.e. quiescence) and cell cycle re-entry was induced by treatment with 100 ng/ml MCSF. Macrophages were trypsinized and collected by centrifugation for 5 minutes at 300g. After fixation in 80% ethanol for at least 1 h at −20°C, cells were incubated for at least 30 minutes with 50 μg/mL propidium iodide containing 0.25 mg/mL RNAse A (both from Sigma). Labelled cells were analyzed in a BD FACSCANTO flow cytometer with FACSDiva software (BD Biosciences) and DNA histograms were fitted into cell cycle distributions using ModFit 3.0 software (Verity Software House). Macrophage proliferation was also assessed by evaluating 5-bromo-2’-deoxyuridine (BrdU) incorporation into BMDM after synchronization in G0 phase by MCSF deprivation and induction of cell-cycle re-entry by treatment with 100 ng/ml MCSF together with 30 μM BrdU (Sigma) for 16h. To detect BrdU incorporation, BMDM were fixed in ethanol at −20oC for 20 min and subjected to acid treatment (10 mM citrate buffer pH 6.0). After neutralization, samples were then blocked with 5% goat serum in PBS-0.1% Tween, and incubated with anti-BrdU primary antibody (ThermoFisher), followed by Alexa Fluor 488-conjugated goat anti-mouse IgG secondary antibody (ThermoFisher). Additional information on these antibodies, including antibody dilutions, can be found in Supplementary Table 10. Nuclei were counterstained with DAPI. Images were captured on a Nikon Eclipse microscope with Nikon AR 4.30.02 (b925) 64bits software, and analyzed using Fiji ImageJ software and the StartDist plugin.
Gene expression analysis by quantitative real-time PCR (qPCR), mRNA sequencing and Western Blot
Total RNA from cultured macrophages or aortic arch tissue was isolated using Trizol reagent and RNeasy kits (QIAGEN). RNA was reverse transcribed with High-Capacity cDNA Reverse Transcription Kit (Applied Bosystems) and qPCR was performed with SYBR® Green PCR Master Mix (Applied Biosystems) in a AB7900 Real time PCR system. Results were analyzed with the ΔΔCt method. The average of 36B4 and β-actin was used as reference for normalization. Primer sequences are listed in Supplementary Table 11.
Libraries for mRNA sequencing were prepared using the SMART-Seq v4 Ultra Low Input RNA Kit (Clontech-Takara). Full-length cDNAs were processed using a Nextera XT DNA Library Preparation Kit (Illumina). Quality control was performed using a High Sensitivity DNA ScreenTape Assay with a 4200 TapeStation System (Agilent). Libraries were then multiplexed in an equimolar pool and sequenced using a NextSeq 550 Platform (Illumina). Single-end unstranded libraries (75bp) were first subjected to a quality check FastQC v0.11.8 and aligned to the reference genome assembly mm10 with the following settings (STAR --outFilterMultimapNmax 20 --alignSJoverhangMin 8 –alignSJDBoverhangMin 1 --outFilterMismatchNmax 999 --outFilterMismatchNoverReadLmax 0.04 --alignIntronMin 20 -- alignIntronMax 1000000 --alignMatesGapMax 1000000 --outSAMattributes NH HI NM MD).
RNA-seq quality assessment was performed using RSeQC v3.0.1 tool. Differential expression analysis was assessed with edgeR v3.30.0 R Bioconductor package (PMID: 19910308) and only genes with a cut-off expression > 1 CPM in at least three samples were included in the analysis. TMM method was applied to normalize gene counts and glmQLFTest function was used to perform differential analysis, with a minimum cut-off of absolute fold change ≥ 1.5 and FDR ≤0.05. A complete list of differentially expressed genes and statistical information (pValue and FDR) for each condition is available at Gene Expression Omnibus (GEO: GSE184420). Functional enrichment analysis was performed using Ingenuity Pathway Analysis software (IPA v01-13; Qiagen). GOplot representations were used to combine and integrate the expression data with the results of the functional analysis44.
For Western Blot analysis of protein levels, protein extracts from cultured macrophages were obtained using ice-cold lysis buffer (20mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM NA2EDTA, 1 mM EGTA, 1% Triton) supplemented with protease and phosphatase inhibitors (Roche Applied Science). Equal amounts of protein lysates were resolved by SDS-PAGE and the following antibodies were used for immunoblotting: anti-p53 (Cell Signaling) and HRP-conjugated anti-α tubulin (Abcam). Additional information on these antibodies, including antibody dilutions, can be found in Supplementary Table 10. An ImageQuant LAS 4000 biomolecular imaging system with Image Quant LAS4000 software (GE Healthcare) was used for image acquisition, and Fiji ImageJ software was used for band densitometric analysis. p53 protein levels were normalized to α-tubulin.
Analysis of modified lipoprotein uptake and efferocytosis in cultured macrophages
Thioglycolate-elicited peritoneal macrophages were cultured in RPMI medium supplement with antibiotics and 10% FBS. To asses modified LDL uptake, peritoneal macrophage were treated with 5 μg/ml fluorescent acetylated LDL (DiI acLDL, ThermoFisher) for 1h or 3h. Macrophage were then trypsinized, collected and analyzed by flow cytometry using a BD FACSymphony flow cytometer with BD FACSDiva software (BD Biosciences). Data were analyzed with FlowJo Software (Tree Star). To assess uptake of apoptotic cells (i.e. efferocytosis), thymuses from C57Bl/6J mice were collected, homogenized, filtered through a 70μm cell strainer and labeled fluorescently with 5 μM CellTrace Violet (ThermoFisher). Thymocyte apoptosis was induced by exposure to UV light for 10 minutes. 1 or 2 hours later, apoptotic thymocyte were added to macrophage cultures at a 5:1 ratio to allow apoptotic cell engulfment by macrophages. After 1h, macrophages were washed extensively, collected by trypsinization and stained with FITC-conjugated anti-CD11b (Invitrogen) and PE-conjugated anti-F4/80 antibodies (Tonbo Bioscience). Additional information on these antibodies, including antibody dilutions, can be found in Supplementary Table 10. Samples were analyzed by flow cytometry using a BD FACSCanto flow cytometer with BD FACSDiva software (BD Biosciences). Data were analyzed with FlowJo Software (Tree Star).
Analysis of neutrophil phenotype and function
Neutrophil phenotypes analyses and ROS quantification were performed in blood samples extracted from experimental mice and treated with RBC hypotonic lysis buffer (0.15M NH4Cl, 0.01M KHCO3 and 0.01M EDTA in water). For ROS quantification, cells were stimulated with 133 nM of phorbol 12-myristate 13-acetate (PMA) for 20 min or PBS as control, and then stained with 5 mM Dihydrorhodamine 123 (DHR123, Thermo Fisher) for 20 min, and with BUV737-conjugated anti-Ly6G antibody (BD Biosciences), PE-conjugated anti-CD11b antibody (Tonbo Biosciences), PE-Cy7-conjugated anti-CD45.1 antibody (BioLegend) and APC-conjugated anti-CD45.2 antibody (BioLegend) for 15 min. For surface marker analysis, samples were incubated with BUV737-conjugated anti-Ly6G antibody (BD Biosciences), PE-conjugated anti-CD62L antibody (eBioscience), PerCP-Cy5.5-conjugated anti-CXCR2 antibody (BioLegend), BV510-conjugated anti-CD11b antibody (BioLegend), PECy7-conjugated anti-CD101 antibody (ThermoFisher), AlexaFluor 647-conjugated anti-CD54 antibody (BioLegend), APC-Cy7-conjugated anti-CD45 antibody (BioLegend) and FITC-conjugated anti-CD45.2 antibody (BioLegend) for 15 min. Additional information on these antibodies, including antibody dilutions, can be found in Supplementary Table 10. Dead cells were excluded from analysis by DAPI staining. Samples were analyzed in a BD FACSymphony Cytometer with BD FACSDiva software (BD Biosciences) for data acquisition. Analysis was performed using FlowJo software (Tree Star).
NETosis was assessed in cryopreserved aortic root sections from hematopoietic chimeric mice, and cryopreserved lung sections from mice subjected to transfusion-related acute lung injury as positive control45. After fixing in 4% PFA for 15 min at RT, samples were blocked with 5% BSA, 1% normal goat serum, 5% FBS and 0.1% triton-x 100 in PBS in a humid chamber for 1h followed by an incubation with antibodies against citrullinated histone 3 (Abcam), CD31 (Thermo Fisher) and biotinylated-MPO (R&D) overnight at 4°C. Sections were then incubated with goat anti-rabbit-Alexa 568 (Life Technologies), goat anti-hamster-Alexa-647 (Jackson Immunoresearch), Alexa-488 conjugated Streptavidin (BioLegend) and DAPI (Life Technologies), and mounted with Mowiol 4-88 (Mw 31,000, Sigma). Additional information on antibodies, including antibody dilutions, can be found in Supplementary Table 10. Imaging of NETs was performed using a Leica SP5 multi-line inverted confocal microscope with 63x magnification and LAS-AF 2.7.3. build 9723 software. Images were analyzed using Fiji ImageJ software (NIH). An event was considered positive for NETosis when it was triple stained (MPO, cit H3, and DAPI).
Statistical analysis of data in experimental studies
Data are shown as mean ± SEM unless otherwise stated. Statistical significance of differences in experiments with two groups and only one variable was assessed by unpaired Student’s t tests (with Welch correction for unequal variance when appropriate) or Mann-Whitney tests. Differences in experiments with more than one independent variable were evaluated by two-way analysis of variance (ANOVA) with post-hoc Sidak’s or Tukey’s multiple comparison tests. All statistical tests were performed using GraphPad Prism software (GraphPad Software Inc.).
Extended Data
Extended Data Figure 1. Association of CHIP with blood counts among individuals without prevalent hematologic malignancy in the UK Biobank (n=37,657).

Blood counts were acquired at time of blood draw for whole exome sequencing. a) Association of CHIP and Large CHIP with normalized blood counts (SD). Associations are adjusted for age, age2, sex, smoking status, and the first ten principal components of genetic ancestry. b) Association of CHIP variant allele frequency (VAF) with blood counts (in units of 10^9 cells/L). The gray horizontal dotted lines reflect average counts across non-CHIP carriers. The vertical black dotted line reflects the cutoff VAF for Large CHIP (VAF>0.1). Error bands reflect the standard error of a generalized additive model with integrated smoothness fit to the data. CHIP = clonal hematopoiesis of indeterminate potential; VAF = variant allele fraction
Extended Data Figure 2. Association of CHIP (a) and VAF (b) with incident hematologic malignancy among individuals without prevalent hematological malignancy in the UK Biobank (n=37,657).

Associations are adjusted for age, age2, sex, smoking status, Townsend deprivation index, and the first ten principal components of genetic ancestry. (a) Error bars are centered at the HR and show the 95% CI for estimates. (b) Error bands reflect the standard error of a generalized binomial additive model with integrated smoothness fit to the data. CHIP = clonal hematopoiesis of indeterminate potential; VAF = variant allele fraction
Extended Data Figure 3. Epidemiological causal inference analysis for CHIP on incident peripheral artery disease in the UK Biobank.

a) Propensity scores by CHIP and Large CHIP status in the UKB (n=37,657). b) Propensity score adjustment and stabilized inverse probability treatment weighting (IPTW) for the CHIP and Large CHIP association with incident PAD in the UKB. Error bars are centered at the HR and show the 95% CI for estimates. CHIP = clonal hematopoiesis of indeterminate potential; VAF = variant allele fraction; PAD = peripheral artery disease
Extended Data Figure 4. Association of Large CHIP (VAF>10%) with incident pan-arterial atherosclerosis, combined across peripheral artery disease, coronary artery disease, aneurysms, chronic and acute mesenteric ischemia, cerebral atherosclerosis, and renal artery stenosis.

Error bars are centered at the HR and show the 95% CI for estimates. CHIP = clonal hematopoiesis of indeterminate potential; VAF = variant allele fraction
Extended Data Figure 5. Association of a) CHIP and b) Large CHIP genes with incident pan-arterial atherosclerosis, combined across peripheral artery disease, coronary artery disease, aneurysms, chronic and acute mesenteric ischemia, cerebral atherosclerosis, and renal artery stenosis.

Error bars are centered at the HR and show the 95% CI for estimates. CHIP = clonal hematopoiesis of indeterminate potential; VAF = variant allele fraction
Extended Data Figure 6. Effects of experimental p53-deficient CHIP on atherosclerosis development in Ldlr−/− mice.
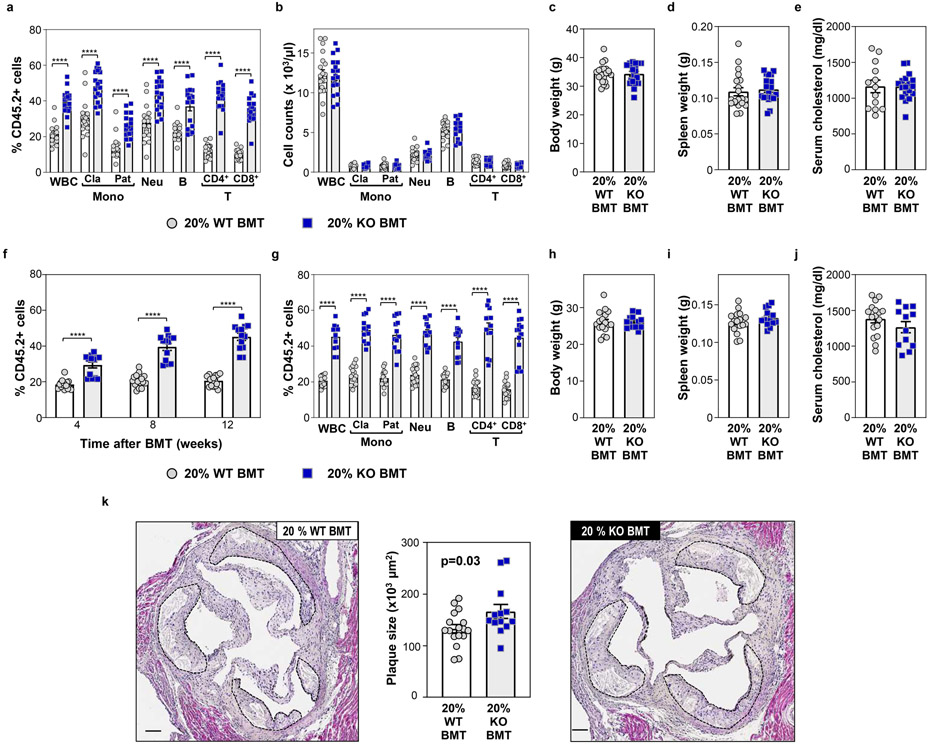
a-e) 20% KO-BMT male mice (n=19 mice) and 20% WT-BMT controls (n=20 mice) were fed a high-fat/high-cholesterol (HF/HC) diet for 9 weeks, starting 4 weeks after BMT. a) Percentage of CD45.2+ cells in different white blood cell (WBC) lineages in peripheral blood, evaluated by flow cytometry. Two-tailed unpaired t-tests were used for statistical analysis (mean±SEM, ****p<0.0001). b) Absolute counts of main WBC sub-populations in peripheral blood, evaluated by flow cytometry (mean±SEM). c) Body weight (mean±SEM). d) Spleen weight (mean±SEM). e) Total cholesterol level in serum, evaluated by enzymatic methods (mean±SEM). f-k) 20% KO-BMT female mice and 20% WT-BMT controls were fed a high-fat/high-cholesterol (HF/HC) diet for 9 weeks, starting 4 weeks after BMT. f) Percentage of CD45.2+ cells in white blood cells at different timepoints, evaluated by flow cytometry. A two-way ANOVA with Sidak’s multiple comparison test was used for statistical analysis (mean±SEM,****p<0.0001). g) Percentage of CD45.2+ cells in different WBC lineages in peripheral blood after 9 weeks on HF/HC diet (13 weeks post-BMT), evaluated by flow cytometry. Two-tailed unpaired t-tests were used for statistical analysis (mean±SEM, n=16 20% WT-BMT mice, n=13 20% KO-BMT mice, ****p<0.0001). h) Body weight (mean±SEM, n=17 20% WT-BMT mice, n=13 20% KO-BMT mice). i) Spleen weight (mean±SEM, n=17 20% WT-BMT mice, n=13 20% KO-BMT mice). j) Total cholesterol level in serum, evaluated by enzymatic methods (mean±SEM, n=17 20% WT-BMT mice, n=12 20% KO-BMT mice) k) Aortic root plaque size. A two-tailed unpaired t-test was used for statistical analysis (mean±SEM, n=17 20% WT-BMT mice, n=13 20% KO-BMT mice). Representative images of hematoxylin and eosin-stained sections are shown; atherosclerotic plaques are delineated by dashed lines. Scale bars, 100 μm.
Extended Data Figure 7. No effect of p53-deficient CHIP on plaque lipid content or aortic expression of the proinflammatory cytokines IL-1b and IL-6 or the NLRP3 inflammasome.

a) Aortic arch samples were obtained from HF/HC-fed 20% KO-BMT male mice and 20% WT-BMT controls, and gene expression was analyzed by qPCR (mean±SEM, n=11 20% WT-BMT mice, n=10 20% KO-BMT mice). b) 20% KO-BMT male mice and 20% WT-BMT controls were fed a high-fat/high-cholesterol (HF/HC) diet for 9 weeks. Plaque lipid content was analyzed through Oil Red O (ORO) staining of cryostat sections (mean±SEM, n=12 20% WT-BMT mice, n=15 20% KO-BMT mice). Representative images of ORO-stained sections are shown; atherosclerotic plaques are delineated by dashed lines. Scale bars, 200 μm.
Extended Data Figure 8. Increased cell proliferation in conditions of p53-deficient CHIP.
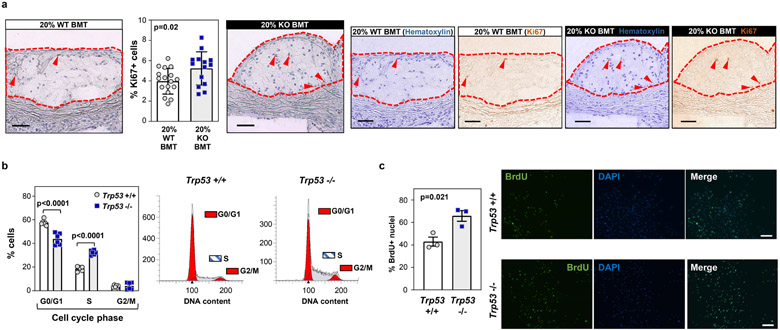
a) 20% KO-BMT female mice and 20% WT-BMT controls were fed a high-fat/high-cholesterol (HF/HC) diet for 9 weeks, starting 4 weeks after BMT. Plaque cell proliferation was estimated based on immunohistochemical staining of the Ki-67 proliferation marker (mean±SEM, n=17 20% WT-BMT mice, n=14 20% KO-BMT mice). A two-tailed unpaired t-test was used for statistical analysis. Representative images of Ki-67-stained sections are shown; color deconvolution was applied to show separately the staining of hematoxylin (nuclei) and Ki-67. Atherosclerotic plaques are delineated by dashed lines. Scale bars, 50 μm. b) Cell cycle phase distribution of cultured Trp53−/− and +/+ bone marrow-derived macrophages proliferating asynchronously in the presence of 100 ng/ml MCSF, evaluated by propidium iodide staining of cellular DNA content and flow cytometry (mean±SEM, n=6 Trp53+/+ mice, n=6 Trp53−/− mice). A two-way ANOVA with Sidak’s multiple comparison test was used for statistical analysis. c) Analysis of the proliferation of cultured Trp53−/− and +/+ bone marrow-derived macrophages through immunostaining of BrdU incorporation into the DNA. Quiescent G0-synchronized macrophages were treated with MCSF to induce proliferation (mean±SEM, n=3 Trp53+/+ mice, n=3 Trp53−/− mice). A two-tailed unpaired t-test was used for statistical analysis. A representative experiment is shown; three separate experiments were conducted. Scale bars, 100 μm.
Extended Data Figure 9. Transcriptomic profiling of MCSF-stimulated p53-deficient macrophages.

An 18h-treatment with MCSF was used to induce cell cycle entry and progression in quiescent G0-synchronized Trp53−/− (KO) and +/+ (WT) murine macrophages in culture (n=3 per genotype). mRNA sequencing was used for transcriptomic profiling. a) Heatmap of differentially expressed genes with fold change (FC)≥1.5. b) Functional categories enriched in the set of genes differentially expressed between Trp53−/− and +/+ macrophages, based on Ingenuity Pathway Analysis (cancer-related pathways are not shown). c) Heatmap of the most upregulated genes within selected functional categories that are enriched in genes differentially expressed in Trp53−/− macrophages with fold change≥1.5. d) GoPlot of selected functional categories (right hand-side) and the logFC values of the most differentially expressed genes included in these categories (left hand-side).
Extended Data Fig. 10. Association of CHIP and Large CHIP (variant allele fraction>10%) with PAD in the UKB (N=37,657).

under 1) unadjusted, 2) sparsely adjusted, and 3) fully adjusted models, where sparsely adjusted refers to the following covariates: age, age2, sex, smoking status, Townsend deprivation index, and the first ten principal components of genetic ancestry, and the fully adjusted model additionally includes normalized BMI, prevalent hypertension, hyperlipidemia, and type 2 diabetes as covariates. Given the minimal difference between the sparsely adjusted and fully adjusted model, the sparsely adjusted model was moved forward for use in analysis. Error bars are centered at the HR and show the 95% CI for estimates.
Supplementary Material
Acknowledgments
D.K. is supported by the Veterans Administration, Award 1IK2BX005759-01. P.N. is supported by a Hassenfeld Scholar Award from the Massachusetts General Hospital, grants from the National Heart, Lung, and Blood Institute (R01HL1427, R01HL148565, and R01HL148050), and from Fondation Leducq (TNE-18CVD04). S.M.Z is supported by the NIH National Heart, Lung, and Blood Institute (1F30HL149180-01) and the NIH Medical Scientist Training Program Training Grant (T32GM136651). A.G.B. is supported by a Burroughs Wellcome Fund Career Award for Medical Scientists, a NIH Director Early Independence Award (DP5-OD029586) and a NHLBI BioData Catalyst Fellowship (OT3 HL147154-01). J.P.P is supported by a John S LaDue Memorial Fellowship. K.P. is supported by NIH grant 5-T32HL007208-43. P.L. receives funding support from the National Heart, Lung, and Blood Institute (1R01HL134892), the American Heart Association (18CSA34080399), the RRM Charitable Fund, and the Simard Fund. J.J.F. is supported by grants RYC-2016-20026 from the Spanish “Ministerio de Ciencia e Innovación” (MICIN/AEI /10.13039/501100011033” and “ESF Investing in your future”), a 2019 Leonardo Grant for Researchers and Cultural Creators from the BBVA Foundation, the European Research Area Network on Cardiovascular Diseases CHEMICAL (grant AC19/00133, funded by Instituto de Salud Carlos III and co-funded by European Union, ERDF, "A way to make Europe"), and the Leducq Foundation (TNE-18CVD04). A.G.B. is supported by NIH grant DP5 OD029586, a Burroughs Wellcome Fund Career Award for Medical Scientists, and a Pew-Stewart Scholar for Cancer Research award, supported by the Pew Charitable Trusts and the Alexander and Margaret Stewart Trust. C.G. is supported by the European Research Area Network on Cardiovascular Diseases CHEMICAL. A.H. is supported by the Leducq Foundation (TNE-18CVD04). K.P is supported by NIH T32 grant - 5T32HL007208. A.N. received funding from the Knut and Alice Wallenberg Foundation (KAW 2017.0436). C.M.B. is supported by NIH grant number R01-HL148050. C.J.G. is supported by NIH/NCI K08CA263555. P.L. receives funding support from the National Heart, Lung, and Blood Institute (1R01HL134892 and 1R01HL163099-01), the American Heart Association (18CSA34080399), the RRM Charitable Fund, and the Simard Fund. M.A-P is supported by grant FPU18/02913 funded by MCIN/AEI /10.13039/501100011033 and by “ESF Investing in your future”. The project leading to these results also received funding from “la Caixa” Foundation (ID 100010434), including fellowship code LCF/BQ/DR19/11740022 (to A.A-C) and agreement HR17-00267. Confocal microscopy was conducted at the Microscopy & Dynamic Imaging Unit at CNIC, ICTS-ReDib, co-funded by MCIN/AEI /10.13039/501100011033 and FEDER “Una manera de hacer Europa” (#ICTS-2018-04-CNIC-16). The CNIC is supported by the Instituto de Salud Carlos III (ISCIII), the MICIN and the Pro CNIC Foundation, and is a Severo Ochoa Center of Excellence (grant CEX2020-001041-S funded by MICIN/AEI/10.13039/501100011033). We thank R. Moro for assistance with figure preparation, and F. Sánchez-Cabo and M. Gómez for assistance with bioinformatics analysis of RNA-seq data. S.M.D. is supported by the Veterans Administration, award IK2-CX001780. We also thank participants and staff of the UK Biobank and Mass General Brigham Biobank. UK Biobank analyses were conducted using Application 7089.
Abbreviations:
- CHIP
clonal hematopoiesis of indeterminate potential
- PAD
peripheral artery disease
Footnotes
Competing Interest Statement
D.K. is a scientific advisor for and accepts consulting fees from Bitterroot Bio, Inc. P.N. reports grant support from Amgen, Apple, AstraZeneca, Boston Scientific, and Novartis, spousal employment and equity at Vertex, consulting income from Apple, AstraZeneca, Novartis, Genentech / Roche, Blackstone Life Sciences, Foresite Labs, and TenSixteen Bio, and is a scientific advisor board member and shareholder of TenSixteen Bio and geneXwell, all unrelated to this work. P.L. is an unpaid consultant to, or involved in clinical trials for Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Norvo Nordisk, Novartis, Pfizer, and Sanofi-Regeneron. Dr. Libby is a member of the scientific advisory board for Amgen, Caristo Diagnostics, Cartesian Therapeutics, CSL Behring, DalCor Pharmaceuticals, Dewpoint Therapeutics, Eulicid Bioimaging, Kancera, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Moderna, Novartis, PlaqueTec, TenSixteen Bio, Soley Thereapeutics, and XBiotech, Inc. P.L.'s laboratory has received research funding in the last 2 years from Novartis. P.L. is on the Board of Directors of XBiotech, Inc. He has a financial interest in Xbiotech, a company developing therapeutic human antibodies, in TenSixteen Bio, a company targeting somatic mosaicism and clonal hematopoiesis of indeterminate potential (CHIP) to discover and develop novel therapeutics to treat age-related diseases, and in Soley Therapeutics, a biotechnology company that is combining artificial intelligence with molecular and cellular response detection for discovering and developing new drugs, currently focusing on cancer therapeutics. P.L.’s interests were reviewed and are managed by Brigham and Women’s Hospital and Mass General Brigham in accordance with their conflict-of-interest policies. B.L.E. has received research funding from Celgene, Deerfield, Novartis, and Calico and consulting fees from GRAIL. He is a member of the scientific advisory board and shareholder for Neomorph Inc., TenSixteen Bio, Skyhawk Therapeutics, and Exo Therapeutics. A.G.B. is a paid advisor and holds equity in TenSixteen Bio. S.M.D. is received a research support via the University of Pennsylvania from RenalytixAI and Novo Nordisk. The other authors do not report any disclosures.
Data Availability
UKB individual-level data are available by request via application (https://www.ukbiobank.ac.uk). Individual-level MGBB data are available from https://personalizedmedicine.partners.org/Biobank/Default.aspx, only to Partners HealthCare investigators with appropriate approval from the Partners Institutional Review Board (IRB). RNA-seq data are available at Gene Expression Omnibus (GEO: GSE184420). The present article includes all other data generated or analyzed during this study.
Code Availability
Our CHIP calling Terra pipeline using Mutect2 (v1) is available here (https://app.terra.bio/#workspaces/terra-outreach/CHIP-Detection-Mutect2). R-scripts for the observational epidemiologic associations are available at https://github.com/mzekavat/CHIP_PAD .
References
- 1.Song P, et al. Global, regional, and national prevalence and risk factors for peripheral artery disease in 2015: an updated systematic review and analysis. Lancet Glob Health 7, e1020–e1030 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Conte MS, et al. Global vascular guidelines on the management of chronic limb-threatening ischemia. J Vasc Surg 69, 3S–125S.e140 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaiswal S, et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. New England Journal of Medicine 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaiswal S, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. New England Journal of Medicine 377, 111–121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie M, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nature medicine 20, 1472–1478 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Genovese G, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bick AG, et al. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation 141, 124–131 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pascual-Figal DA, et al. Clonal Hematopoiesis and Risk of Progression of Heart Failure With Reduced Left Ventricular Ejection Fraction. J Am Coll Cardiol 77, 1747–1759 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Yu B, et al. Supplemental Association of Clonal Hematopoiesis With Incident Heart Failure. J Am Coll Cardiol 78, 42–52 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhattacharya R, et al. Clonal Hematopoiesis Is Associated With Higher Risk of Stroke. Stroke 53, 788–797 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bick AG, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 586, 763–768 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klarin D, et al. Genome-wide association study of peripheral artery disease in the Million Veteran Program. Nature medicine 25, 1274–1279 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Denny JC, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol 31, 1102–1110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuster JJ, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science (New York, N.Y.) 355, 842–847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Visconte V, M ON & H JR Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features. Cancers (Basel) 11(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boettcher S, et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science (New York, N.Y.) 365, 599–604 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bondar T & Medzhitov R p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell 6, 309–322 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 4, 37–48 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.TeKippe M, Harrison DE & Chen J Expansion of hematopoietic stem cell phenotype and activity in Trp53-null mice. Exp Hematol 31, 521–527 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Sano S, et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1beta/NLRP3 Inflammasome. J Am Coll Cardiol 71, 875–886 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaiswal S, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med 377, 111–121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuster JJ, et al. TET2-Loss-of-Function-Driven Clonal Hematopoiesis Exacerbates Experimental Insulin Resistance in Aging and Obesity. Cell reports 33, 108326 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abplanalp WT, et al. Clonal Hematopoiesis-Driver DNMT3A Mutations Alter Immune Cells in Heart Failure. Circ Res 128, 216–228 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Sinha SK, et al. Local M-CSF (Macrophage Colony-Stimulating Factor) Expression Regulates Macrophage Proliferation and Apoptosis in Atherosclerosis. Arterioscler Thromb Vasc Biol 41, 220–233 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuster JJ Clonal hematopoiesis and cardiovascular disease in cancer patients and survivors. Thromb Res 213 Suppl 1, S107–S112 (2022). [DOI] [PubMed] [Google Scholar]
- 26.Boesten LS, et al. Macrophage p53 controls macrophage death in atherosclerotic lesions of apolipoprotein E deficient mice. Atherosclerosis 207, 399–404 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Fuster JJ, et al. Control of cell proliferation in atherosclerosis: insights from animal models and human studies. Cardiovasc Res 86, 254–264 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Guevara NV, Kim HS, Antonova EI & Chan L The absence of p53 accelerates atherosclerosis by increasing cell proliferation in vivo. Nat Med 5, 335–339 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Mercer J, Figg N, Stoneman V, Braganza D & Bennett MR Endogenous p53 protects vascular smooth muscle cells from apoptosis and reduces atherosclerosis in ApoE knockout mice. Circ Res 96, 667–674 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Merched AJ, Williams E & Chan L Macrophage-specific p53 expression plays a crucial role in atherosclerosis development and plaque remodeling. Arterioscler Thromb Vasc Biol 23, 1608–1614 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Sanz-Gonzalez SM, et al. Increased p53 gene dosage reduces neointimal thickening induced by mechanical injury but has no effect on native atherosclerosis. Cardiovasc Res 75, 803–812 (2007). [DOI] [PubMed] [Google Scholar]
- 32.van Vlijmen BJ, et al. Macrophage p53 deficiency leads to enhanced atherosclerosis in APOE*3-Leiden transgenic mice. Circ Res 88, 780–786 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Robbins CS, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med 19, 1166–1172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Min KD, Polizio AH, Kour A, Thel MC & Walsh K Experimental ASXL1-Mediated Clonal Hematopoiesis Promotes Inflammation and Accelerates Heart Failure. J Am Heart Assoc 11, e026154 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heyde A, et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell 184, 1348–1361 e1322 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fidler TP, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 592, 296–301 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bycroft C, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smoller JW, et al. An eMERGE Clinical Center at Partners Personalized Medicine. Journal of personalized medicine 6(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Hout CV, et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 586, 749–756 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaiswal S, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu P, et al. Mapping ICD-10 and ICD-10-CM Codes to Phecodes: Workflow Development and Initial Evaluation. JMIR Med Inform 7, e14325 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zekavat SM, et al. Hematopoietic mosaic chromosomal alterations and risk for infection among 767,891 individuals without blood cancer. medRxiv, 2020.2011.2012.20230821 (2020). [Google Scholar]
- 43.Hernan MA, Brumback B & Robins JM Marginal structural models to estimate the causal effect of zidovudine on the survival of HIV-positive men. Epidemiology 11, 561–570 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Walter W, Sánchez-Cabo F & Ricote M GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics 31, 2912–2914 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Adrover JM, et al. Programmed "disarming" of the neutrophil proteome reduces the magnitude of inflammation. Nat Immunol 21, 135–144 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
UKB individual-level data are available by request via application (https://www.ukbiobank.ac.uk). Individual-level MGBB data are available from https://personalizedmedicine.partners.org/Biobank/Default.aspx, only to Partners HealthCare investigators with appropriate approval from the Partners Institutional Review Board (IRB). RNA-seq data are available at Gene Expression Omnibus (GEO: GSE184420). The present article includes all other data generated or analyzed during this study.
Our CHIP calling Terra pipeline using Mutect2 (v1) is available here (https://app.terra.bio/#workspaces/terra-outreach/CHIP-Detection-Mutect2). R-scripts for the observational epidemiologic associations are available at https://github.com/mzekavat/CHIP_PAD .