

SUMMARY
Major histocompatibility complex (MHC) class II molecules present products of lysosomal proteolysis to CD4+ T cells. While extracellular antigen uptake is considered to be the main source of MHC class II-ligands, a few intracellular antigens have been described to gain access to MHC class II loading after macroautophagy. However, the general relevance and efficacy of this pathway is unknown. Here we demonstrate constitutive autophagosome formation in MHC class II positive cells, including dendritic, B and epithelial cells. The autophagosomes continuously fuse with multivesicular MHC class II-loading compartments (MIICs). This pathway is of functional relevance, since targeting of the Influenza Matrix Protein 1 (MP1) to autophagosomes via fusion to the autophagosome-associated protein Atg8/LC3 led to strongly enhanced MHC class II presentation to CD4+ T cell clones. We suggest that macroautophagy constitutively and efficiently delivers cytosolic proteins for MHC class II presentation and can be harnessed for improved helper T cell stimulation.
Keywords: MHC class II, macroautophagy, CD4+ T cells, Influenza matrix protein 1, Atg8/LC3
INTRODUCTION
The T cells of the adaptive immune system monitor all body cells for the presence of pathogenic proteins. For this purpose, peptides generated by the proteasome are presented on MHC class I molecules and recognized by CD8+ T cells, whereas products of lysosomal degradation are presented on MHC class II molecules and recognized by CD4+ T cells (Bryant and Ploegh, 2004; Cresswell et al., 2005; Trombetta and Mellman, 2005). Antigens presented on MHC class II are typically exogenous proteins that are endocytosed by the antigen presenting cell (APC) or endogenous proteins that reside in the secretory system. However, analysis of peptides eluted from MHC class II molecules has revealed that a significant proportion of natural MHC class II ligands (up to 20%) are derived from cytosolic and nuclear proteins (Chicz et al., 1993; Dengjel et al., 2005; Dongre et al., 2001; Friede et al., 1996; Muntasell et al., 2004). Furthermore, it was shown that CD4+ T cells can recognize cytosolic and nuclear antigens after endogenous processing. This pathway for CD4+ T cell recognition was first described by Long and colleagues, who showed that cytosolic measles virus and influenza A virus antigens can be endogenously processed for MHC class II presentation (Gueguen and Long, 1996; Jacobson et al., 1989; Jaraquemada et al., 1990). Subsequently, endogenous MHC class II presentation has been described for other viral antigens (Chen et al., 1998; Paludan et al., 2005), self antigens (Brazil et al., 1997; Lich et al., 2000), model antigens (Nimmerjahn et al., 2003; Qi et al., 2000), as well as tumor antigens (Dorfel et al., 2005; Zeng et al., 2001). Therefore, antigens that are topologically isolated from the endosomal system can gain access to the MHC class II antigen presentation pathway and broaden the repertoire of MHC class II ligands.
Different processing pathways have been discussed to contribute to endogenous MHC class II antigen presentation (reviewed in (Lechler et al., 1996; Schmid and Münz, 2005)). Recently, macroautophagy was shown to deliver cytosolic and nuclear antigens onto MHC class II molecules (Brazil et al., 1997; Dengjel et al., 2005; Dorfel et al., 2005; Nimmerjahn et al., 2003; Paludan et al., 2005; Zhou et al., 2005). Of these, the Epstein-Barr virus (EBV) nuclear antigen 1 (EBNA1) was the first pathogen-derived antigen that was found to be presented on MHC class II molecules of EBV-transformed B cell lines after macroautophagic degradation (Paludan et al., 2005). Autophagy consists of at least three intracellular protein degradation pathways found ubiquitously in eukaryotic cells, chaperone-mediated macroautophagy, microautophagy and macroautophagy (Shintani and Klionsky, 2004). Both chaperone-mediated autophagy and macroautophagy have been implicated in MHC class II presentation of cytosolic and nuclear antigens (reviewed in (Deretic, 2006)). During chaperone-mediated autophagy, LAMP-2a with the assistance of one cytosolic and one lysosomal heat shock protein 70 family member imports cytosolic substrates in a signal peptide-dependent fashion directly into lysosomes (Majeski and Dice, 2004). In contrast, during macroautophagy, cytoplasmic material, including organelles and protein aggregates, is sequestered into double membrane-coated autophagosomes, which subsequently fuse with endosomes and lysosomes to form so-called amphisomes or autolysosomes (Yorimitsu and Klionsky, 2005). In these fusion compartments, the sequestered contents of autophagosomes are then broken down by lysosomal hydrolases and the degradation products are recycled by the cell.
So far, MHC class II presentation to CD4+ T cells after macroautophagic degradation has only been described for one model antigen (neomycin phosphotransferase II}(Nimmerjahn et al., 2003), one self antigen (Complement C5) (Brazil et al., 1997), one tumor antigen (MUC-1) (Dorfel et al., 2005) and one viral antigen (EBNA1) (Paludan et al., 2005). Therefore, we wondered whether macroautophagy is a general and efficient pathway for MHC class II presentation of intracellular antigens in a variety of antigen presenting cells. In order to address this question, we quantified macroautophagy in MHC class II-positive human cells, analyzed the overlap of autophagosomes with MHC class II loading compartments and tested whether targeting of antigens for macroautophagic degradation leads to enhanced MHC class II presentation. Here we show that macroautophagy is a constitutively active pathway in all MHC class II-positive cells, including dendritic, B and epithelial cells. Furthermore, we show that MHC class II compartments continuously receive input from the cytoplasm via macroautophagy and, accordingly, the targeting of antigens to autophagosomes leads to enhanced MHC class II presentation and thus CD4+ T cell stimulation.
RESULTS
Autophagosomes are constitutively turned over by lysosomal proteases in human epithelial cell lines
To test whether MHC class II-positive human cells exhibit constitutive macroautophagy, we first quantified the level of macroautophagy in human epithelial cell lines. Epithelial cells readily up-regulate MHC class II molecules in response to inflammatory cytokines both in vitro and in vivo (Reith and Mach, 2001). Since they have only limited endocytic potential, we wondered if these cell lines might rely on endogenous degradation pathways, such as macroautophagy, to generate peptide ligands for their MHC class II molecules. To quantify macroautophagy, we made use of the specific autophagosome marker Atg8/LC3. LC3 is an ubiquitin-like protein that is covalently coupled via its C-terminus to a phospholipid in the newly forming inner and outer autophagosomal membranes and thus is specifically incorporated into autophagosomes (Kabeya et al., 2000). After fusion of autophagosomes with endosomes or lysosomes, intraluminal LC3 is rapidly degraded by lysosomal proteases. The more autophagosomes that are formed, the more LC3 is degraded in autolysosomes and therefore, lysosomal turnover of LC3 is a good measure for macroautophagic activity (Tanida et al., 2005).
To visualize the lysosomal turnover of LC3 in human epithelial cells by fluorescence microscopy, we transfected cell lines derived from different organs [HaCat (skin), HeLa (cervix), MDAMC (breast), 293 (kidney)] with a GFP-LC3 fusion construct. GFP-LC3 reporter constructs have been used previously to visualize autophagosomes in transgenic mice and cultured cells (Mizushima, 2005), and it has been shown that overexpression of GFP-LC3 does not alter the macroautophagic activity ((Mizushima et al., 2004) and our unpublished data). GFP-LC3-transfected cell lines were treated with the lysosomal acidification inhibitor chloroquine (CQ) to block lysosomal proteolysis and thus visualize the accumulation of GFP-LC3 in autolysosomes. In the steady state (no CQ), most cells had only a few GFP-LC3-labeled autophagosomes (typically 0–5) and only a small fraction of cells had >10 GFP-LC3-positive vesicles (Fig. 1A, left). However, in all cell lines examined, GFP-LC3 strongly accumulated in cytosolic vesicles after 10 hours of CQ-treatment (Fig. 1A, right), suggesting that large numbers of GFP-LC3 labeled autophagosomes had formed and fused with lysosomes during the 10-hour observation period. The accumulation of brightly GFP-LC3 labelled vesicles could already be observed 2 hours after CQ-treatment (Fig. S1A), in good agreement with the rapid degradation kinetics of these vesicles. The accumulation of GFP-LC3+ vesicles upon CQ treatment was dependent on macroautophagy, since siRNA-mediated knockdown of atg12, a gene essential for autophagosome formation, completely abrogates accumulation of these vesicles (Fig. 1B).
Figure 1. Constitutive autophagosome formation in human epithelial cell lines under nutrient-rich conditions.

(A) Human epithelial cell lines HaCat (keratinocyte), HeLa (cervical carcinoma), MDAMC (breast carcinoma), and 293 (kidney) were stably transfected with a GFP-LC3 reporter construct. To assess turnover of GFP-LC3 in endosomal/lysosomal compartments, cells were treated with 50 μM chloroquine for 10 h (+CQ) or were left untreated (no CQ). Cells were fixed, stained with DAPI nucleic acid stain and analyzed by confocal microscopy. Scale bars: 20 μm. Representative fields from one experiment out of three are shown.
(B) MDAMC cells stably expressing GFP-LC3 were either mock transfected or with siRNA duplexes specific for lamin A/C or atg12. After 2 days, cells were treated with 50 μM CQ for 6 h (+CQ) or were left untreated (no CQ), stained with DAPI and examined in an epifluorescence microscope. One of two experiments is shown.
(C) Human epithelial cell lines (HaCat, HeLa, MDAMC, and 293) were treated for 10 h with 50μM CQ (+) or were left untreated (−). Whole cell lysates were run on a 12% SDS-PAGE gel and LC3-I and –II were visualized by anti-LC3 Western blotting. Actin blots show that CQ-treatment did not affect general protein levels. One of three experiments is shown.
(D) HaCat cells were treated with 50 μM CQ for 0, 1, 2, 10 or 24 h and gradual accumulation of LC3-II was visualized by anti-LC3 Western blot. Anti-actin blot controls for sample loading. One of two experiments is shown.
To extend our results with GFP-LC3 to endogenous LC3, we took advantage of the fact that autophagosome-associated LC3 (called LC3-II) and free cytosolic LC3 (called LC3-I) can be distinguished by their apparent molecular weights in SDS-PAGE gel electrophoresis (16 and 18 kD, respectively) and thus can be quantified separately in anti-LC3 Western blots (Kabeya et al., 2000). The same four epithelial cell lines were cultured in the presence or absence of the lysosomal protease inhibitor CQ for 10 hours and accumulation of LC3-II was quantified by immunoblotting. In all cell lines, autophagosome-associated LC3-II strongly accumulated upon CQ-treatment (Fig. 1C), demonstrating that LC3-II labeled autophagosomes were constitutively degraded in endosomes/lysosomes over the course of 10 hours. As shown in Fig. 1D for the HaCat cell line, cellular LC3-II levels were already increased 1 hour after CQ-treatment and gradually accumulated after longer treatment times, confirming that autophagosomes are being produced continuously. Density quantification of Western blots revealed that LC3-II accumulated 5-fold (HaCat and MDAMC), 15-fold (HeLa) and 30-fold (293) after 10 hours of CQ-treatment (data not shown). LC3-II accumulated to similar levels when cells were treated with inhibitors of lysosomal cathepsins for 10 hours (Fig. S1B). Taken together, these experiments confirm that macroautophagy is a constitutively active process in all human epithelial cell lines analyzed. Although macroautophagy can be associated with nutritional deprivation (Codogno and Meijer, 2005), the gradual accumulation of LC3 demonstrates that autophagosomes continuously deliver LC3 for lysosomal degradation, i.e., that macroautophagy is constitutively active in human epithelial cell lines under nutrient-rich conditions.
Macroautophagy is a constitutively active process in professional antigen-presenting cells, including dendritic cells
Next, we sought to analyze the macroautophagy level of professional antigen-presenting cells (APCs) that are constitutively MHC class II-positive, such as B cells and dendritic cells. Dendritic cells (DCs) are the most efficient professional APCs and exist in two functionally and phenotypically distinct states, immature and mature (Mellman and Steinman, 2001). Immature DCs continuously circulate through tissues and lymphoid organs to present antigens to T cells for tolerance induction (Steinman et al., 2003). In contrast, mature DCs have an exceptional capacity to initiate adaptive and innate immune responses. Given the crucial role of DCs in both tolerance and immunity, we were wondering whether macroautophagy might contribute to MHC class II presentation in these cells.
To quantify the macroautophagy level in human B cells and dendritic cells, we visualized the turnover of lentivirally delivered GFP-LC3 in EBV-transformed B lymphoblastoid cell lines (LCL) and in monocyte-derived immature and mature DCs. In all three cell types, GFP-LC3-labeled autophagosomes strongly accumulated after treatment with CQ for 10 h (Fig. 2A), indicating constitutive autophagosome turnover. While LCL only contained up to 10 autophagosomes after CQ exposure, as previously observed (Paludan et al., 2005), especially immature DCs accumulated as many of GFP-LC3+ vesicles as the epithelial cell lines. Furthermore, LC3 immunoblots confirmed that endogenous LC3 is continuously degraded by lysosomal proteases in human B cell lines (Fig. 2B), in freshly isolated CD14+ monocytes as well as in monocyte-derived immature and mature dendritic cells (Fig. 2C). These experiments demonstrated that macroautophagy is a steady-state process not only in epithelial cells, which present antigens on MHC class II only upon immune activation, but also in professional APCs. Furthermore, constitutive macroautophagy is not restricted to transformed human cell lines, but is also a feature of primary human cells, as demonstrated for primary monocytes and dendritic cells.
Figure 2. Macroautophagy is a constitutive process in professional antigen-presenting cells (APCs), including dendritic cells.
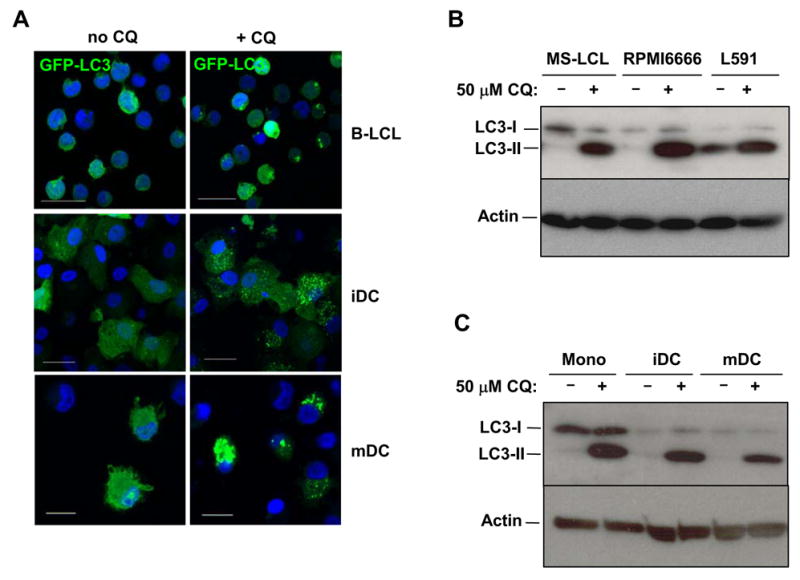
(A) To assess lysosomal turnover of GFP-LC3 in professional APCs, an EBV-transformed B lymphocyte cell line (B-LCL) stably expressing GFP-LC3 and GFP-LC3-expressing immature and mature DCs (iDC and mDC) were treated with 50 μM chloroquine for 10 h (+CQ) or were left untreated (no CQ). Cells were fixed, stained with DAPI and analyzed by confocal microscopy. Scale bars: 20 μm. Representative fields from one experiment out of two are shown.
(B and C) Human B cell lines [MS-LCL (EBV-transformed B lymphocyte cell line), RPMI6666 and L591 (Hodgkin's lymphoma cell lines)] (B), CD14+ monocytes (Mono), immature DCs (iDC) and LPS-matured DCs (mDC) (C) were treated for 10 h with 50 μM CQ (+) or were left untreated (−). Whole cell lysates were run on 12% SDS-PAGE gels and LC3-I and –II were visualized by anti-LC3 Western blotting. Actin blots show that CQ-treatment did not affect general protein levels. One of three experiments is shown.
GFP-LC3 colocalizes with markers of MHC class II loading compartments in IFN-γ-treated human epithelial cell lines
To test whether autophagosomes fuse with MHC class II loading compartments (MIICs), we examined by confocal microscopy whether the autophagosome marker GFP-LC3 would colocalize with markers of MIICs. MIICs have been characterized as conventional late endosomal compartments that in addition to late endosomal/lysosomal markers, such as LAMP-1 and –2, contain the components for MHC class II loading, namely MHC class II and the peptide-loading chaperone HLA-DM (Trombetta and Mellman, 2005).
We initially focused our analysis on epithelial cells, since they might rely heavily on endogenous MHC class II antigen processing due to their limited endocytic potential compared to classical antigen presenting cells like dendritic cells. We confirmed that most of our human epithelial cell lines, with the exception of 293 cells, expressed MHC class II molecules after IFN-γ treatment (data not shown). In the human cell lines used in this study, IFN-γ treatment did not lead to a detectable upregulation of macroautophagy, as determined by immunoblot (Fig. S2), although we cannot exclude that the IFN-γ treatment slightly influenced the macroautophagy level, in addition to inducing MHC class II-positive compartments. For colocalization analysis, cells were treated with IFN-γ, transiently transfected with the GFP-LC3 reporter construct, and stained with antibodies specific for the MIIC markers MHC class II, HLA-DM and LAMP-2 in MDAMC (Fig. 3A) and HaCat cells (data not shown). As shown in Fig. 3A, MHC class II positive compartments frequently colocalized with GFP-LC3. When we quantified colocalization of MHC class II and HLA-DM with GFP-LC3 using the LSM510 software's profile tool, which overlays the intensity profiles along a cross section through a cell, we found that in double-positive cells, 58% (± 10%) of MHC class II+ and 52% (± 20%) of HLA-DM+ compartments contained GFP-LC3.
Figure 3. The autophagosome marker GFP-LC3 colocalizes with markers of MHC class II loading compartments in human epithelial cell lines and dendritic cells.

(A) MDAMC cells were treated with 200 U/ml IFN-γ, transiently transfected with a GFP-LC3 reporter construct and 48 h later stained with antibodies to MHC class II, HLA-DM, LAMP-2 and DAPI for DNA content. Colocalization of GFP-LC3 with MIIC markers was analyzed by confocal microscopy. Scale bars: 10 μm. Representative cells from one experiment out of three are shown.
(B) Same as (A), except that 50 μM chloroquine (CQ) was present during the last 10 h of the culture. Scale bars: 10 μm. Representative cells from one experiment out of three are shown.
(C) Colocalization of GFP-LC3 and MHC class II molecules in electron-dense multivesicular compartments. Untreated (I and II) or CQ-treated (III and IV) MDAMC cells stably expressing GFP-LC3 and MHC class II-positive due to IFN-γ induction were fixed in 4% paraformaldehyde and cut into 80 nm-thin cryosections. Sections were labeled with an HLA-DR-specific antiserum and 10 nm protein A-Gold (PAG10) and antibody-PAG complexes were irreversibly fixed with glutaraldehyde. Subsequently, sections were labeled with a GFP-specific antibody and 15 nm protein A-Gold (PAG15) and were analyzed by electron microscopy. As a control, PAG10 and PAG15 were interchanged and were shown to produce the same labeling pattern (I/II vs. III/IV). Insets from panels I and III are shown at higher magnification in panels II and IV, respectively. Representative fields from one experiment out of three are shown.
To assess the proportion of MIICs that showed no colocalization with GFP-LC3 due to degradation of the autophagosome marker protein, we performed the same experiments on CQ-treated MDAMC and HaCat cells. Colocalization of GFP-LC3 with MHC class II, HLA-DM and LAMP-2 was more pronounced under these conditions (Fig. 3B and data for HaCat not shown). Colocalization analysis with the LSM 510 profile tool showed that after CQ treatment, colocalization increased to 86% (± 9%) for MHC class II+ and to 71% (± 12%) for HLA-DM+ vesicles (Fig. 4B). This demonstrates that the majority of MHC class II loading compartments obtain input from autophagosomes in human epithelial cell lines.
Figure 4. GFP-LC3 is degraded in MHC class II loading compartments of dendritic cells.

(A) GFP-LC3-expressing immature DCs were treated with 50 μM chloroquine for 10 h (+CQ). Cells were stained with an MHC class II-specific antibody and DAPI and colocalization of GFP-LC3 with MHC class II was analyzed by confocal microscopy. Scale bar: 10 μm. Representative cells from one experiment out of three are shown.
(B) Same experiment as in (A) was performed with mature DCs. In the majority of mature DCs MHC class II was mainly localized at the cell surface, but a subset of cells had intracellular MHC class II compartments (white arrow). Scale bar: 10 μm. Representative cells from one experiment out of three are shown.
GFP-LC3 and MHC class II colocalize in electron-dense multivesicular compartments
To identify GFP-LC3/MHC class II double-positive compartments by electron microscopy, we prepared ultrathin cryosections of untreated or CQ-treated, stably GFP-LC3 transfected MDAMC cells, stained them with antibodies specific for HLA-DR and GFP, and applied antibodies labeled with 10 and 15 nm protein A-Gold particles. In both, untreated and CQ-treated cells, the two antibodies strongly labeled large (1–2 μm), electron-dense, multivesicular compartments (Fig. 3C). Other organelles, such as nuclei and mitochondria, were mostly gold-negative, however some GFP-LC3 staining was observed in the cytosol and MHC class II staining could be seen on the ER/Golgi and at the cell membrane (Fig. S3A). The morphology of the double-labeled compartments was very similar in untreated and CQ-treated cells, but they were found much more frequently in CQ-treated cells (data not shown) and some of them displayed the characteristic swollen phenotype of lysosomal compartments under chloroquine treatment (Fig. S3B). At higher magnification, the presence of electron-dense material, numerous small vesicles and sometimes larger lipid membranes became apparent (Fig. 3C, panels II and IV). Both GFP-LC3 and MHC class II molecules were often found in close proximity to each other on intraluminal lipid membranes. This suggests that autophagosomes frequently fuse with MHC class II compartments, giving rise to multivesicular compartments that contain both MHC class II molecules and LC3 on internal membranes.
GFP-LC3 is degraded in MHC class II loading compartments of dendritic cells
After having observed an overlap of autophagosomes with MHC class II loading compartments in human epithelial cell lines, we wondered whether we could observe the same colocalization in professional APCs, most notably dendritic cells. To address this question, we transfected CD14+ monocytes with a lentiviral GFP-LC3 reporter construct, generated immature and mature DCs and stained them with an MHC class II-specific antibody. Since DCs are constitutively MHC class II-positive, no IFN-γ treatment was necessary for these experiments.
As shown in Fig. 4A, MHC class II compartments of immature DCs frequently contained the autophagosome marker GFP-LC3 after 10 hours of CQ-treatment. Colocalization analysis with the LSM510 profile tool showed that 41% (±7%) of MHC class II-labeled compartments were positive for GFP-LC3. In the majority of mature DCs, MHC class II molecules were mainly localized at the cell surface (Fig. 4B) and therefore the overlap with autophagosomes was minimal. However, in a subset of cells that still had some intravesicular MHC class II staining, GFP-LC3 was frequently localized within these MIICs after CQ treatment (Fig. 4B, white arrow). In the absence of the lysosomal acidification inhibitor CQ, GFP-LC3 was mainly present in the cytosol and only very few GFP-LC3+ vesicles could be observed in both immature and mature DCs (see Fig. 2A). Therefore, no colocalization analysis could be performed for untreated DCs. However, the accumulation of GFP-LC3 in MIICs of CQ-treated immature and mature DCs showed that autophagosomes feed into the MHC class II pathway not only in epithelial cell lines but also in professional APCs, namely dendritic cells.
GFP-LC3 minimally colocalizes with markers of early endosomes or MHC class I loading compartments
To determine if autophagosomes selectively fuse with MIICs, we analyzed the overlap of GFP-LC3 with markers of other endocytic compartments, specifically early endosomes (positive for early endosomal antigen, EEA1) and recycling endosomes (positive for transferrin receptor, TR). As shown in Fig. 5A for the MDAMC cell line, GFP-LC3 did not show a pronounced overlap with either EEA1 or transferrin receptor, even in the presence of chloroquine. When we quantified colocalization of EEA1 with GFP-LC3, colocalization was low in untreated MDAMC cells (9%), but slightly increased after chloroquine treatment (to 26%) (Fig. 5B). The difference between GFP-LC3 colocalization with MHC class II or HLA-DM versus EEA1 was statistically significant in the presence and absence of CQ (p < 0.001) (Fig. 5B). In HaCat cells, the overlap of GFP-LC3 with EEA1 or transferrin receptor was also minimal (data not shown). Furthermore, in IFN-γ treated cells, GFP-LC3 rarely entered early or recycling endosomes (data not shown).
Figure 5. GFP-LC3 minimally colocalizes with markers of early endosomes or MHC class I loading compartments.

(A) MDAMC cells were transiently transfected with a GFP-LC3 reporter construct and 36 h later treated with 50 μM chloroquine for 10 h. Cells were stained with antibodies to early endosomal antigen (EEA1) or transferrin receptor (TR) and DAPI and analyzed by confocal microscopy. Scale bars: 10 μm. Representative cells from one experiment out of two are shown.
(B) Quantitative analysis for colocalization of GFP-LC3 with MHC class II, HLA-DM and EEA1 in untreated or CQ-treated MDAMC cells. Data represent means from 10–15 cells from one representative experiment out of two. Error bars indicate standard deviations. P-values from homocedastic, one-tailed student’s T test statistics are shown.
(C) As in (A), except that cells were stained with an MHC class I-specific antibody.
Macroautophagy has been implicated in the presentation of intracellular antigens on MHC class II, but does not seem to influence MHC class I presentation (Nimmerjahn et al., 2003; Paludan and Münz, 2003). To further address this issue, we analyzed the overlap of GFP-LC3 with MHC class I-molecules. As expected, MHC class I was mainly found in perinuclear ER/Golgi regions and on the plasma membrane and did not colocalize with the more peripherally distributed GFP-LC3-positive vesicles (Fig. 5C). Together, our data suggest that autophagosomes mainly fuse with MIICs in MHC class II positive cells, but only rarely with early/recycling endosomes or MHC class I loading compartments.
Cytosolic/nuclear antigens can be targeted for macroautophagic degradation by fusion to Atg8/LC3
The observation that autophagosomes continuously fuse with MHC class II loading compartments suggests that this pathway might deliver cytosolic antigens efficiently for MHC class II presentation to CD4+ T cells. To test this hypothesis, we investigated whether the targeting of a cytosolic antigen for macroautophagy would lead to enhanced CD4+ T cell recognition. For this purpose, we generated a fusion construct of the Influenza matrix protein 1 with the autophagosome marker protein Atg8/LC3, reasoning that the LC3 portion of such a fusion protein should target the antigen to macroautophagic membranes and subsequently degradation in MIICs. We then stably expressed the MP1-LC3 fusion protein or the MP1 wild-type protein (Fig. 6A) in the human epithelial cell lines HaCat and MDAMC. Western blot analysis showed that the antigens were expressed in both cell lines and had the expected molecular weights (MP1: 28 kD; MP1-LC3: 43 kD) (Fig. 6B). Notably, the MP1-LC3 fusion protein was present at slightly lower levels than MP1 in both cell lines. In order to test whether the fusion protein would indeed localize to autophagosomes, we coexpressed GFP-LC3 and the MP1-constructs in MDAMC cells (Fig. 6C) and HaCat cells (data not shown) and analyzed their colocalization by confocal microscopy. As expected, wild-type MP1 was homogenously distributed in the cytosol and nucleus, and this pattern did not change after CQ-treatment (Fig. 6C, top rows). In contrast, the MP1-LC3 fusion protein showed punctate cytosolic staining, which overlapped with GFP-LC3-positive autophagosomes (Fig. 6C, bottom rows). Furthermore, after CQ-treatment for 10 hours, GFP-LC3 and MP1-LC3 accumulated in the same cytosolic vesicles, demonstrating that the LC3 tag indeed targets the influenza matrix protein 1 to autophagosomes for subsequent degradation by lysosomal proteases.
Figure 6. Targeting of influenza A matrix protein 1 (MP1) to autophagosomes by fusion to Atg8/LC3.
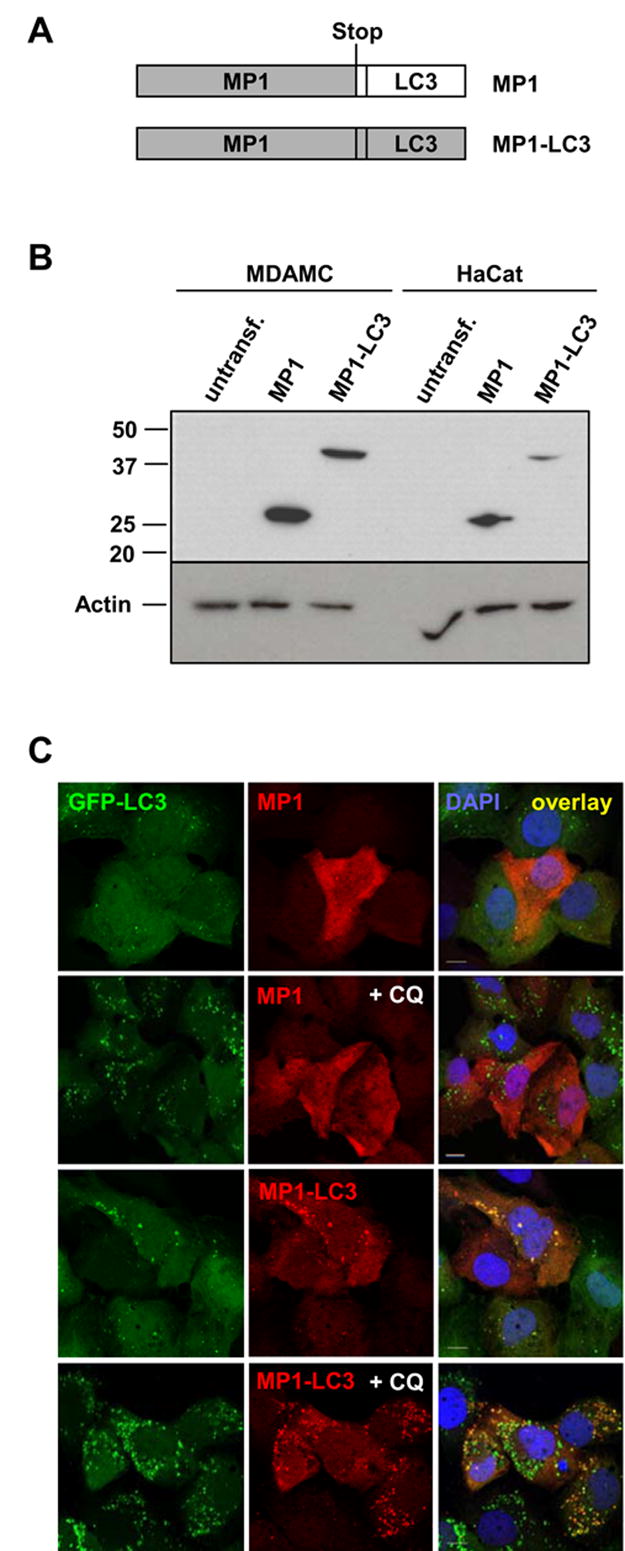
(A) Schematic diagram of the two constructs encoding for MP1 and MP1-LC3, respectively. The influenza A virus matrix protein 1 (MP1) coding sequence was fused to the N-terminus of the human LC3 sequence, either with or without a stop codon at the 3′ end of MP1.
(B) HaCat and MDAMC cell lines were stably transfected with lentiviral MP1 and MP1-LC3 constructs and protein expression was analyzed by Western blot with anti-MP1 antiserum. Actin blot shows equal protein loading.
(C) MDAMC cells stably expressing GFP-LC3 were infected with lentivirus encoding for MP1 or MP1-LC3. To inhibit degradation of autophagosome substrates in lysosomes, cells were treated with 50 μM CQ for 10 h (+CQ). Cells were stained with anti-MP1 antiserum and DAPI and analyzed by confocal microscopy. Scale bars: 10 μm. Representative fields from one experiment out of two are shown.
Targeting of antigens for macroautophagic degradation leads to enhanced CD4+ T cell recognition
To test the hypothesis that targeting of cytosolic/nuclear antigens for macroautophagic degradation via LC3 fusion leads to enhanced MHC class II presentation, we analyzed the recognition of MP1- versus MP1-LC3-expressing target cells by MP1-specific CD4+ T cell clones. For this purpose, we generated MP1-specific CD4+ T cell clones (Fig. S4) from a healthy lab donor that was HLA-DR and –DQ matched to the HaCat cell line, so that IFN-γ treated HaCat cells could be used as target cells. To analyze the effect of the LC3 fusion on MHC class I presentation, we also generated an HLA-A2-restricted CD8+ T cell clone from the same donor (Fig. S4), which then could be tested for recognition of HLA-A2-positive MDAMC target cells. Furthermore, autologous EBV-transformed B-LCLs and monocyte-derived DCs from the same donor could be used as target cells for both CD4+ and CD8+ T cell clones.
To assess how well the two different forms of MP1 could be presented on MHC class II, we measured IFN-γ secretion of three MP1-specific CD4+ T cell clones in response to MP1 or MP1-LC3-expressing target cells (HaCat epithelial cells, B-LCLs or immature/mature DCs). As shown in Fig. 7A, the response of the CD4+ T cell clones (clone 9.26, 10.9 and 11.46) was strongly increased by the LC3 fusion. This effect was seen for all types of target cells, epithelial, B and dendritic cells. While at the lowest ratio of T cell clone to cellular targets (effector to target or E:T ratio) MP1-LC3 typically elicited only 2–4 fold higher IFN-γ production, the difference in IFN-γ secretion was especially pronounced at higher E:T ratios, when the target cells and thus MHC class II-peptide complexes became limiting. At these E:T ratios, IFN-γ secretion by the CD4+ T cell clones in response to MP1-LC3 compared to MP1 was increased 12–17 fold for HaCat epithelial targets, 4–8 fold for B-LCL targets, 5–17 fold for immature DCs and 7–20 fold for mature DCs. Untransfected and GFP-LC3 transfected target cells were not recognized above background (Fig. 7A). While the IFN-γ response to MP1 transfectants of the HaCat cell line never exceeded 30% of the amount secreted upon recognition of the peptide-pulsed HaCat positive control, MP1-LC3 transfectants were able to stimulate up to 95% of the maximal CD4+ T cell recognition achieved with peptide pulsed targets. Mixing experiments demonstrated that MHC class II presentation of MP1 and MP1-LC3 was indeed due to endogenous processing, since the mixing of HLA-matched HaCat cells with mismatched MP1- or MP1-LC3 expressing MDAMC cells did not stimulate any T cell responses (Fig. 7A, upper panel). Furthermore, when MHC class II was not induced on HaCat cells by IFN-γ, MP1- and MP1-LC3-expresssing cells were unable to stimulate CD4+ T cell responses (Fig. 7A, upper panel), confirming that the presentation was MHC class II-restricted. In addition, MHC class II surface staining showed that equal MHC class II surface levels were present on all MP1- and MP1-LC3-expressing target cells (data not shown), demonstrating that the enhanced recognition of MP1-LC3 was not due to an enhanced MHC class II expression level.
Figure 7. Fusion of MP1 to LC3 enhances CD4+ T cell recognition, while leaving CD8+ T cell recognition unaffected.

(A) MP1-specific CD4+ T cell clones 9.26, 10.9 and 11.46 were stimulated at various effector to target cell (ET) ratios with different MP1 or MP1-LC3 expressing target cells: HaCat cells (IFN-γ-treated to induce MHC class II), CM-LCL or immature/mature DCs. The next day, IFN-γ in culture supernatants was measured by ELISA to assess MHC class II presentation of MP1. As a positive control, target cells were pulsed with cognate peptide (+pept.) and as negative controls, untransfected and GFP-LC3-transfected target cells were used. Error bars indicate standard deviations and P-values for paired, one-tailed student’s T test statistics across all E:T ratios are shown. For each target cell type, one representative clone out of three is shown and experiment was performed twice.
(B) As in (A), but MP1-specific CD8+ T cell clone 9.2 was used for all experiments. Target cells were MP1 or MP1-LC3 expressing MDAMC cells (IFN-γ-treated or untreated), CM-LCL or immature/mature DCs. Error bars indicate standard deviations. One of two experiments is shown.
To assess the effect of the LC3 fusion on MHC class I presentation, we analyzed the IFN-γ response of an MP1-specific CD8+ T cell clone to MP1- and MP1-LC3-expressing target cells (MDAMC epithelial cells, B-LCLs or immature/mature DCs). For all types of target cells and across all E:T ratios, similar amounts of IFN-γ were secreted by CD8+ T cells in response to MP1- and MP1-LC3 expressing targets (Fig. 7B). This suggests that the LC3 fusion does not impair MHC class I presentation and both constructs give rise to similar amounts of defective ribosomal products (DRiPs), which are then efficiently processed for CD8+ T cell recognition (Yewdell et al., 2003). This was observed for both IFN-γ-treated MDAMC target cells and untreated MDAMC cells, although MHC class I presentation seemed to be slightly enhanced by the IFN-γ treatment, which is consistent with an enhanced MHC class I processing machinery (Trombetta and Mellman, 2005). Taken together, our data suggest that targeting of cytosolic/nuclear antigens for macroautophagic degradation via LC3 fusion can strongly increase CD4+ T cell recognition, without impairing CD8+ T cell recognition.
Autophagosome targeting depends on covalent coupling of LC3 to the autophagosome membrane via Gly120 and on macroautophagic delivery to MHC class II loading compartments
In order to determine if coupling to the autophagosomal membrane was crucial for antigen targeting to enhance MHC class II presentation, we mutated the amino acid Gly120 of LC3 to Ala in our fusion construct (MP1-LC3(G120A)). MP1-LC3(G120A) failed to localize to GFP-LC3+ autophagosomes (Fig. 8A) and was not preferentially targeted for MHC class II presentation. Influenza MP1 specific CD4+ T cell recognition of MP1-LC3(G120A) was similar to MP1 after transfection into HaCat cells (Fig. 8B). All constructs were expressed to similar levels in HaCat and MDAMC cells (data not shown) and elicited comparable Influenza MP1 specific CD8+ T cell stimulation (Fig. 8B). These data suggest that the ubiquitin-like conjugation of LC3 to the autophagosomal membrane is crucial for LC3 mediated targeting to enhance MHC class II presentation.
Figure 8. Autophagosome targeting depends on covalent coupling of LC3 to the autophagosome membrane via Gly120.

(A) MDAMC cells stably expressing GFP-LC3 were infected with lentivirus encoding for MP1, MP1-LC3 or MP1-LC3(G120A). To visualize autophagosomes, cells were treated with 50 μM CQ for 10 h (+CQ) and were stained with anti-MP1 antiserum and DAPI and analyzed by confocal microscopy. Scale bars: 10 μm.
(B) The MP1-specific CD4+ T cell clone 11.46 or the MP1-specific CD8+ T cell clone 9.2 were stimulated at various effector to target cell (ET) ratios with target cells expressing either MP1, MP1-LC3 or MP1-LC3(G120). The next day, IFN-γ in culture supernatants was measured by ELISA to assess presentation of MP1 on MHC class II or I, respectively. Error bars indicate standard deviations and P-values for paired, one-tailed student’s T test statistics across all E:T ratios are shown. One of two experiments is shown.
To further confirm that macroautophagy is required for targeting of LC3 fusion antigens to MHC class II loading compartments, we analyzed colocalization of MP1-LC3 with MHC class II with and without atg12-specific RNA interference (Fig. S5). In cells transfected with control siRNA, MP1-LC3 colocalized substantially with MHC class II in cytosolic vesicles of IFN-γ-treated MDAMC cells. In contrast, in cells transfected with atg12-specific siRNA, MP1-LC3 was diffusely distributed and did not colocalize with punctuate MHC class II compartments any more. These experiments suggest that LC3 fusion antigens enter MHC class II loading compartments primarily via macroautophagy.
Taken together, the two ubiquitin-like conjugation system, LC3-lipid and Atg12-Atg5, are required for targeting of LC3 fusion proteins for MHC class II loading and enhanced MHC class II presentation, since mutation of the residue involved in LC3 conjugation to the autophagic membrane as well as atg12-specific RNA interference inhibit this pathway.
DISCUSSION
Several recent studies have implicated different autophagic pathways in endogenous MHC class II antigen processing (Brazil et al., 1997; Dengjel et al., 2005; Dorfel et al., 2005; Nimmerjahn et al., 2003; Paludan et al., 2005; Zhou et al., 2005), but none of them evaluated the degree to which autophagy is constitutively active in MHC class II positive antigen presenting cells (APCs) and how efficient these pathways are for antigen delivery to the MHC class II loading compartment. We provide evidence in our study that constitutively MHC class II positive APCs, such as B cell lines, monocytes and dendritic cells, as well as epithelial cell lines with IFN-γ-inducible MHC class II expression display significant levels of steady-state macroautophagy. We found that autophagosomes constitutively fuse with MHC class II loading compartments in epithelial and dendritic cells, and that by targeting this pathway via fusion to the autophagosome marker Atg8/LC3, we could strongly increase MHC class II presentation and CD4+ T cell recognition of Influenza MP1. These data suggest that macroautophagy constitutively and efficiently delivers cytosolic antigens for MHC class II presentation.
In line with our findings, analysis of GFP-LC3 transgenic mice has shown that in some murine tissues, e.g. thymus and kidney epithelium, macroautophagy occurs actively even under nutrient-rich conditions (Mizushima et al., 2004). Our study confirms these results and extends constitutive macroautophagy to cell types relevant to the immune system, including B cells, monocytes, dendritic cells and MHC class II-positive epithelial cells. Constitutive macroautophagy might represent a previously underestimated pathway for MHC class II presentation of intracellular proteins, especially in non- or poorly phagocytic MHC class II-positive tissues, such as B cells as well as epithelial and tumor cells, which upregulate MHC class II in response to inflammatory cytokines, such as IFN-γ (Reith and Mach, 2001). Interestingly, MHC class II positive epithelial cells have been found to be surrounded by cytolytic CD4+ T cells at sites of inflammation (Yawalkar et al., 2001), suggesting immune surveillance by CD4+ T cells of these tissues in vivo. Furthermore, cytolytic anti-viral CD4+ T cells have been characterized in both mouse and man ex vivo (Appay et al., 2002; Jellison et al., 2005) and have been demonstrated to control viral infections (Fu et al., 2004; Paludan et al., 2002; Sparks-Thissen et al., 2004). Therefore, non- or poorly phagocytic cell types might depend on endogenous antigen processing routes, such as macroautophagy, to load antigen onto their MHC class II molecules for immune surveillance of these tissues by CD4+ T cells.
In dendritic cells, the most proficient antigen-presenting cells of the immune system, MHC class II presentation after macroautophagy might contribute to the induction of tolerance and immunity by immature and mature DCs, respectively. Although immature DCs are very adept in picking up exogenous antigens, such as dying cells and environmental proteins, to induce tolerance to self and innocuous antigens (reviewed in (Steinman and Nussenzweig, 2002)) or immunity after activation (reviewed in (Banchereau and Steinman, 1998)), intracellular proteins must also be processed onto MHC class II of DCs to comprehensively tolerize against DC specific antigens, and efficiently prime naïve CD4+ T cells after direct infection of DCs. The latter mechanism might be especially important for noncytolytic pathogens that leave infected host cells intact, so that dying cells are not readily available as a source of exogenous antigen. In these situations, macroautophagy might contribute to endogenous MHC class II antigen processing in DCs.
Our analysis by electron micrsocopy suggests that autophagosome content meets MHC class II molecules in multivesicular bodies (Fig. 3C). The ultrastructural features of these MHC class II- and Atg8/LC3-containing compartments are consistent with previous descriptions of MHC class II loading compartments (Li et al., 2005; Trombetta and Mellman, 2005; Zwart et al., 2005). Likewise, colocalization of GFP-LC3 with HLA-DM, a hallmark molecule of the MHC class II loading machinery, suggests that autophagosomes fuse with MIICs (Figure 3B and C). Multivesicular and multilamellar vesicles have been identified as the sites of MHC class II loading and a consensus is emerging that these compartments are late endosomes, which in APCs are equipped with the MHC class II loading machinery (Kleijmeer et al., 1997). Consistent with our findings, autophagosomes have been described to fuse with late endosomes, forming so called amphisomes (Berg et al., 1998; Liou et al., 1997), but the significance of this fusion event for antigen targeting to MHC class II presentation had not been investigated. Our study closes this knowledge gap and implicates fusion of autophagosomes with late endosomal MHC class II loading compartments as a means to continually present cytosolic proteins to CD4+ T cells.
Influenza MP1 could be targeted in our study for enhanced MHC class II presentation by fusion to the autophagosome-associated Atg8/LC3 protein. Although earlier work by Long and colleagues and our experiments with unmodified MP1 reveals access of MP1 to MHC class II presentation by an intracellular route ((Gueguen and Long, 1996) and Fig. 6A), fusion to Atg8/LC3 significantly increased MHC class II presentation of MP1 by up to 20 fold. This increase occurred in spite of the fact that the MP1-LC3 fusion protein was expressed at lower levels than wild-type MP1 after transfection and did not influence surface expression of MHC class II. Therefore, the targeting by macroautophagy might be harnessed to boost MHC class II presentation and CD4+ T cell stimulation of vaccine antigens after intracellular delivery for example via viral vectors. Since strongly enhanced MHC class II presentation after LC3 targeting also occurred in dendritic cells, the most potent activators of naïve T cells, this strategy might efficiently elicit primary CD4+ T cell responses against vaccine antigens. Notably, the Atg8/LC3 fusion protein of MP1 used in this study was also efficiently processed onto MHC class I. Therefore, dendritic cells, which express antigens that are efficiently targeted to autophagosomes via this pathway, would be expected to efficiently prime both CD4+ and CD8+ T cell responses. Since it has been demonstrated that CD4+ T cells in addition to the above outlined direct anti-viral function are essential for the maintenance of protective CD8+ T cell effector functions and memory (Bevan, 2004), improved stimulation of helper T cells could be a valuable component of vaccine development. Particularly in recombinant viral vaccines, targeting of antigens for endogenous MHC class II processing via macroautophagy should be considered.
EXPERIMENTAL PROCEDURES
Cell culture
All human epithelial cell lines were routinely cultured in DMEM with 10% FCS, 2 mM glutamine, 110 μg/ml sodium pyruvate and 2 μg/ml gentamycin. B cell lines and hybridomas were maintained in RPMI-1640 with 10% FCS, + glutamine + gentamycin. Sources of cell lines are included in the Supplemental Data. Monocytes and dendritic cells (DCs) were prepared from PBMCs isolated from leukocyte concentrates (buffy coats) from the New York Blood Center or blood donations from healthy lab donors. Positive selection for CD14-positive monocytes/macrophages was performed using anti-CD14 MicroBeads from Miltenyi Biotec. DCs were generated from CD14-positive cells in RPMI-1640 + 1% single-donor plasma + glutamine + gentamycin. Recombinant human IL-4 (rhIL-4, 500 U/ml) and rhGMCSF (1000 U/ml) were added on day 0, 2, and 4. For maturation, floating immature DCs were transferred to new plates on day 5 and half of the medium was replaced with fresh medium containing proinflammatory cytokines [IL-1β (10 ng/ml), IL-6 (1000 U/ml), TNF-α (10 ng/ml) and PGE2 (1 μg/ml)] or 200 ng/ml lipopolysaccharide (LPS). All cytokines were obtained from R&D Systems, Peprotech, Berlex or Sigma. For lentiviral transduction of DCs, CD14+ monocytes were infected with lentivirus at an MOI of 10 on day 1 after isolation, in the presence of 8μg/ml polybrene and IL-4/GM-CSF and immature/mature DCs were generated as described above.
LC3 fusion constructs and generation of stable transfectants
The cDNA of human MAP1LC3B sequence (NM_022818) was cloned from a human B-LCL by RT-PCR with gene specific primers. The cDNA of Influenza A/WSN/33 matrix protein 1 (MP1) was PCR-amplified from the pCAGGS/MCS-MP1 vector, a gift from Peter Palese, Mount Sinai School of Medicine, New York. A more detailed description of plasmids and lentiviral constructs can be found in the Supplemental Data. HeLa and 293 cell lines stably expressing GFP-LC3 were generated by transient transfection with lipofectamine 2000 (Invitrogen) and subsequent culture in the presence of 500 μg/ml G418. HaCat, MDAMC and CM-LCL cell lines stably expressing GFP-LC3, MP1-LC3 or MP1 were generated by lentiviral infection using MOIs of 10–40.
Knockdown of atg12
The following 21-nt siRNA oligos were used: Atg12 sense: 5′-UCAACUUGCUACUACAUGAUdT; Atg12 antisense: 5′-UCAUGUAGUAGCAAGUUGAUdT (nt. 687–705 of NM_004707). As a control, lamin A/C-specific siRNA from Dharmacon (Lamin sense: 5′-CUGGACUUCCAGAAGAACAdTdT; Lamin antisense: 5′-UGUUCUUCUGGAAGUCCAGdTdT) or firefly luciferase-specific siRNA (GL2 sense: 5′-CGUACGCGGAAUACUUCGAdTdT; GL2 antisense: 5′-UCGAAGUAUUCCGCGUACGdTdT) was used. siRNA duplexes were delivered by transfection with lipofectamine 2000 (Invitrogen) at 30 pmol siRNA + 1.5 μl lipofectamine/well in a 24-well format and effect of knockdown was analyzed after 2–3 days.
Antibodies
The LC3 antiserum was generated by immunizing two rabbits with the N-terminal peptide LC31–15 (MPSEKTFKQRRTFEQR) conjugated to KLH carrier protein (Cocalico Biologicals, Reamstown, PA). Animals were boosted 5 times (2, 3, 7, 11 and 15 weeks after initial inoculation) and then sacrificed to obtain terminal bleeds. Antiserum collected from one rabbit showed good LC3 reactivity by ELISA and Western blot and was used for Western blots at a dilution of 1:2000. Influenza MP1-specific rabbit antiserum was a gift from Ari Helenius, Zürich, Switzerland. Rabbit anti-HLA-DR antiserum C6861 for electron microscopy was a gift from Peter Cresswell, Yale University, New Haven, CT. Rabbit anti-GFP antibody was purchased from Invitrogen-Molecular Probes and anti-βactin antibody from Sigma. Anti-MHC class I and II antibodies for immunocytochemistry were hybridoma supernatants IVA12 and w6/32 from ATCC, anti-HLA-DM (clone MaP-DM1) was from BD Biosciences Pharmingen, anti-LAMP-2 (clone H4B4) from Southern Biotechnology Associates, anti-EEA1 from Santa Cruz Biotech, and anti-transferrin receptor (clone DF 1513) from Sigma. Secondary antibodies for immunohistrochemistry were Rhodamine-Red™-X-(RRX)-conjugated donkey anti-mouse or anti-rabbit or anti-goat IgGs from Jackson ImmunoResearch.
Immunocytochemistry and confocal microscopy
Epithelial cells were grown on microscopy cover glasses in 24 well plates overnight, whereas B cells or dendritic cells were plated onto polylysine-coated cover glasses immediately before the staining procedure. Cells were fixed in 3% paraformaldehyde in PBS for 15 min and permeabilized in 0.1% Triton X-100 in PBS for 5 min. Cells were blocked for 30 min in blocking buffer (from Perkin Elmer's TSA kit) + 0.1% saponin. Primary and secondary antibodies were applied in blocking buffer + 0.1% saponin + 5% normal donkey serum for 30–60 min, followed by three 5 min-washes in PBS + 0.1% saponin. Finally, cells were stained with DAPI nucleic acid stain (0.5 μg/ml, Invitrogen-Molecular Probes) for 1 min and cover glasses were mounted onto microscope slides using Prolong Gold antifade reagent (Invitrogen-Molecular Probes). All steps were carried out at room temperature. Cells were analyzed using an inverted LSM 510 laser scanning confocal microscope (Zeiss Axiovert 200) with a 63 or 100x/1.4 N.A. oil immersion lens using a pinhole diameter of 1 Airy unit. Pictures were taken with the LSM 510 confocal software (Zeiss). Colocalization of markers was quantified using the profile tool of the LSM 510 software. The number of double-positive vesicles compared to the total number of red vesicles was determined in 10–15 double-positive cells/condition.
Electron microscopy and immunolabeling of cryosections
MDAMC cells stably transfected with GFP-LC3 were fixed for 1h at RT with 4% paraformaldehyde (PFA, Electron Microscopy Sciences) in 0.25 M Hepes, pH 7.4, followed by overnight fixation at 4°C in 8% PFA/Hepes. Cells were embedded in 5% gelatin in PBS, small pieces of gelatin pellets were infiltrated overnight at 4°C with 2.3 M sucrose in PBS, mounted onto cryospecimen pins and frozen in liquid nitrogen. Ultrathin sections (80 nm) were cut using a Leica ultracut ultramicrotome with an FCS cryoattachment at −108°C and collected on formvar- and carbon-coated nickel grids using a 1:1 mixture of 2% methyl cellulose (25 centipoises; Sigma) and 2.3 M sucrose in PBS. Cells were quenched with 0.1 M NH4Cl in PBS, blocked in 1% fish skin gelatin (FSG, Sigma) in PBS and subsequently labeled with rabbit anti-HLA-DR antiserum and 10 or 15 nm protein A–gold (purchased from the Department of Cell Biology, University of Utrecht, Netherlands). After fixation in 1% glutaraldehyde and quenching with 0.1 M NH4Cl, the same labeling procedure was repeated for the rabbit anti-GFP antibody. After final fixation in 1% glutaraldehyde, grids were washed 8x in HPLC-grade water. Sections were infiltrated for 10 min on ice with a mixture of 1.8% methylcellulose and 0.5% uranyl acetate (Electron Microscopy Sciences), washed 3x in 0.5% uranyl acetate/1.8% methylcellulose and air-dried. Samples were analyzed in a Tecnai 12 Biotwin (FEI) microscope and pictures were taken using Kodak 4489 film.
T cell clones
Influenza A matrix protein 1 (MP1)-specific T cell clones were generated as previously described (Fonteneau et al., 2001) and outlined briefly in the Supplemental Data. Peptide epitopes of MP1-specific clones were determined in IFN-γ ELISPOT assays using an MP1 peptide library (64 15-mer peptides overlapping by 10 amino acids). Clones were tested for CD4/CD8 expression by staining with anti-CD4-PE and anti-CD8-PE (BD Biosciences Pharmingen) and subsequent analysis by flow cytometry on a FACScalibur instrument (Becton-Dickinson).
T cell assay and IFN-γ ELISA
MP1- and MP-LC3-expressing target cells were cocultured overnight with MP1-specific T cell clones in RPMI-1640 with 5% PHS + glutamine + gentamycin (5% PHS medium) in a 96-well round bottom plate (105 T cells/well + variable target cell numbers). HaCat target cells were treated with 200 U/ml IFN-γ 24h before the coculture to upregulate MHC class II expression, but were washed 3x in DMEM before addition of T cells to remove IFN-γ. After coculture, IFN-γ in culture supernatants was measured using the human IFN-γ ELISA kit from Mabtech according to the manufacturer’s instructions. Recombinant human IFN-γ (Mabtech) at concentrations of 30–2,000 pg/ml was used as a standard. If IFN-γ levels in supernatants exceeded 2000 pg/ml, supernatants were diluted in 5% PHS medium and IFN-γ was remeasured by ELISA.
Statistics
Paired or homocedastic, one-tailed student’s T test statistics were applied where indicated.
Supplementary Material
Acknowledgments
We thank Ralph Steinman for critically reading the manuscript; Rajiv Khanna, Irene Joab, Martina Vockerodt and Dieter Kube for the gift of cell lines; Irina Tcherepanova for the gift of RNA; Markus Landthaler and Thomas Tuschl for the gift of siRNAs; Peter Palese and Jeremy Luban for the gift of DNA vectors; and Ari Helenius and Peter Cresswell for the gift of antibodies.
This work was supported by the Arnold and Mabel Beckman Foundation, the Alexandrine and Alexander Sinsheimer Foundation, the Burroughs Wellcome Fund, the National Cancer Institute (R01CA108609 and R01CA101741) (to C.M.) and in part by a General Clinical Research Center grant (M01-RR00102) from the National Center for Research Resources at the National Institutes of Health (to the Rockefeller University Hospital). D.S. is a recipient of a predoctoral fellowship from the Schering Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Appay V, Zaunders JJ, Papagno L, Sutton J, Jaramillo A, Waters A, Easterbrook P, Grey P, Smith D, McMichael AJ, et al. Characterization of CD4+ CTLs ex vivo. J Immunol. 2002;168:5954–5958. doi: 10.4049/jimmunol.168.11.5954. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Berg TO, Fengsrud M, Stromhaug PE, Berg T, Seglen PO. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem. 1998;273:21883–21892. doi: 10.1074/jbc.273.34.21883. [DOI] [PubMed] [Google Scholar]
- Bevan MJ. Helping the CD8+ T-cell response. Nat Rev Immunol. 2004;4:595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- Brazil MI, Weiss S, Stockinger B. Excessive degradation of intracellular protein in macrophages prevents presentation in the context of major histocompatibility complex class II molecules. Eur J Immunol. 1997;27:1506–1514. doi: 10.1002/eji.1830270629. [DOI] [PubMed] [Google Scholar]
- Bryant P, Ploegh H. Class II MHC peptide loading by the professionals. Curr Opin Immunol. 2004;16:96–102. doi: 10.1016/j.coi.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Chen M, Shirai M, Liu Z, Arichi T, Takahashi H, Nishioka M. Efficient class II major histocompatibility complex presentation of endogenously synthesized hepatitis C virus core protein by Epstein-Barr virus-transformed B-lymphoblastoid cell lines to CD4+ T cells. J Virol. 1998;72:8301–8308. doi: 10.1128/jvi.72.10.8301-8308.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicz RM, Urban RG, Gorga JC, Vignali DA, Lane WS, Strominger JL. Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J Exp Med. 1993;178:27–47. doi: 10.1084/jem.178.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12:1509–1518. doi: 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]
- Cresswell P, Ackerman AL, Giodini A, Peaper DR, Wearsch PA. Mechanisms of MHC class I-restricted antigen processing and cross-presentation. Immunol Rev. 2005;207:145–157. doi: 10.1111/j.0105-2896.2005.00316.x. [DOI] [PubMed] [Google Scholar]
- Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Muller M, Kreymborg K, Altenberend F, Brandenburg J, Kalbacher H, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A. 2005;102:7922–7927. doi: 10.1073/pnas.0501190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V. Autophagy as an immune defense mechanism. Curr Opin Immunol. 2006;18:375–382. doi: 10.1016/j.coi.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Dongre AR, Kovats S, deRoos P, McCormack AL, Nakagawa T, Paharkova-Vatchkova V, Eng J, Caldwell H, Yates JR, 3rd, Rudensky AY. In vivo MHC class II presentation of cytosolic proteins revealed by rapid automated tandem mass spectrometry and functional analyses. Eur J Immunol. 2001;31:1485–1494. doi: 10.1002/1521-4141(200105)31:5<1485::AID-IMMU1485>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Dorfel D, Appel S, Grunebach F, Weck MM, Muller MR, Heine A, Brossart P. Processing and presentation of HLA class I and II epitopes by dendritic cells after transfection with in vitro transcribed MUC1 RNA. Blood. 2005;105:3199–3205. doi: 10.1182/blood-2004-09-3556. [DOI] [PubMed] [Google Scholar]
- Fonteneau JF, Larsson M, Somersan S, Sanders C, Münz C, Kwok WW, Bhardwaj N, Jotereau F. Generation of high quantities of viral and tumor-specific human CD4+ and CD8+ T-cell clones using peptide pulsed mature dendritic cells. J Immunol Methods. 2001;258:111–126. doi: 10.1016/s0022-1759(01)00477-x. [DOI] [PubMed] [Google Scholar]
- Friede T, Gnau V, Jung G, Keilholz W, Stevanovic S, Rammensee HG. Natural ligand motifs of closely related HLA-DR4 molecules predict features of rheumatoid arthritis associated peptides. Biochim Biophys Acta. 1996;1316:85–101. doi: 10.1016/0925-4439(96)00010-5. [DOI] [PubMed] [Google Scholar]
- Fu T, Voo KS, Wang RF. Critical role of EBNA1-specific CD4+ T cells in the control of mouse Burkitt lymphoma in vivo. J Clin Invest. 2004;114:542–550. doi: 10.1172/JCI22053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueguen M, Long EO. Presentation of a cytosolic antigen by major histocompatibility complex class II molecules requires a long-lived form of the antigen. Proc Natl Acad Sci U S A. 1996;93:14692–14697. doi: 10.1073/pnas.93.25.14692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson S, Sekaly RP, Jacobson CL, McFarland HF, Long EO. HLA class II-restricted presentation of cytoplasmic measles virus antigens to cytotoxic T cells. J Virol. 1989;63:1756–1762. doi: 10.1128/jvi.63.4.1756-1762.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaraquemada D, Marti M, Long EO. An endogenous processing pathway in vaccinia virus-infected cells for presentation of cytoplasmic antigens to class II-restricted T cells. J Exp Med. 1990;172:947–954. doi: 10.1084/jem.172.3.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellison ER, Kim SK, Welsh RM. Cutting edge: MHC class II-restricted killing in vivo during viral infection. J Immunol. 2005;174:614–618. doi: 10.4049/jimmunol.174.2.614. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleijmeer MJ, Morkowski S, Griffith JM, Rudensky AY, Geuze HJ. Major histocompatibility complex class II compartments in human and mouse B lymphoblasts represent conventional endocytic compartments. J Cell Biol. 1997;139:639–649. doi: 10.1083/jcb.139.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechler R, Aichinger G, Lightstone L. The endogenous pathway of MHC class II antigen presentation. Immunol Rev. 1996;151:51–79. doi: 10.1111/j.1600-065x.1996.tb00703.x. [DOI] [PubMed] [Google Scholar]
- Li P, Gregg JL, Wang N, Zhou D, O'Donnell P, Blum JS, Crotzer VL. Compartmentalization of class II antigen presentation: contribution of cytoplasmic and endosomal processing. Immunol Rev. 2005;207:206–217. doi: 10.1111/j.0105-2896.2005.00297.x. [DOI] [PubMed] [Google Scholar]
- Lich JD, Elliott JF, Blum JS. Cytoplasmic processing is a prerequisite for presentation of an endogenous antigen by major histocompatibility complex class II proteins. J Exp Med. 2000;191:1513–1524. doi: 10.1084/jem.191.9.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou W, Geuze HJ, Geelen MJ, Slot JW. The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J Cell Biol. 1997;136:61–70. doi: 10.1083/jcb.136.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004;36:2435–2444. doi: 10.1016/j.biocel.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–258. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ . 2005;12(Suppl 2):1535–1541. doi: 10.1038/sj.cdd.4401728. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntasell A, Carrascal M, Alvarez I, Serradell L, van Veelen P, Verreck FA, Koning F, Abian J, Jaraquemada D. Dissection of the HLA-DR4 peptide repertoire in endocrine epithelial cells: strong influence of invariant chain and HLA-DM expression on the nature of ligands. J Immunol. 2004;173:1085–1093. doi: 10.4049/jimmunol.173.2.1085. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F, Milosevic S, Behrends U, Jaffee EM, Pardoll DM, Bornkamm GW, Mautner J. Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur J Immunol. 2003;33:1250–1259. doi: 10.1002/eji.200323730. [DOI] [PubMed] [Google Scholar]
- Paludan C, Bickham K, Nikiforow S, Tsang ML, Goodman K, Hanekom WA, Fonteneau JF, Stevanovic S, Münz C. EBNA1 specific CD4+ Th1 cells kill Burkitt's lymphoma cells. J Immunol. 2002;169:1593–1603. doi: 10.4049/jimmunol.169.3.1593. [DOI] [PubMed] [Google Scholar]
- Paludan C, Münz C. CD4+ T cell responses in the immune control against latent infection by Epstein-Barr virus. Curr Mol Med. 2003;3:341–347. doi: 10.2174/1566524033479771. [DOI] [PubMed] [Google Scholar]
- Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Münz C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- Qi L, Rojas JM, Ostrand-Rosenberg S. Tumor cells present MHC class II-restricted nuclear and mitochondrial antigens and are the predominant antigen presenting cells in vivo. J Immunol. 2000;165:5451–5461. doi: 10.4049/jimmunol.165.10.5451. [DOI] [PubMed] [Google Scholar]
- Reith W, Mach B. The bare lymphocyte syndrome and the regulation of MHC expression. Annu Rev Immunol. 2001;19:331–373. doi: 10.1146/annurev.immunol.19.1.331. [DOI] [PubMed] [Google Scholar]
- Schmid D, Münz C. Immune surveillance of intracellular pathogens via autophagy. Cell Death Differ . 2005;12(Suppl 2):1519–1527. doi: 10.1038/sj.cdd.4401727. [DOI] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks-Thissen RL, Braaten DC, Kreher S, Speck SH, Virgin HW. An optimized CD4 T-cell response can control productive and latent gammaherpesvirus infection. J Virol. 2004;78:6827–6835. doi: 10.1128/JVI.78.13.6827-6835.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci U S A. 2002;99:351–358. doi: 10.1073/pnas.231606698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- Yawalkar N, Hunger RE, Buri C, Schmid S, Egli F, Brand CU, Mueller C, Pichler WJ, Braathen LR. A comparative study of the expression of cytotoxic proteins in allergic contact dermatitis and psoriasis: spongiotic skin lesions in allergic contact dermatitis are highly infiltrated by T cells expressing perforin and granzyme B. Am J Pathol. 2001;158:803–808. doi: 10.1016/S0002-9440(10)64027-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell JW, Reits E, Neefjes J. Making sense of mass destruction: quantitating MHC class I antigen presentation. Nat Rev Immunol. 2003;3:952–961. doi: 10.1038/nri1250. [DOI] [PubMed] [Google Scholar]
- Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ . 2005;12(Suppl 2):1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng G, Wang X, Robbins PF, Rosenberg SA, Wang RF. CD4+ T cell recognition of MHC class II-restricted epitopes from NY-ESO-1 presented by a prevalent HLA DP4 allele: association with NY-ESO-1 antibody production. Proc Natl Acad Sci U S A. 2001;98:3964–3969. doi: 10.1073/pnas.061507398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Li P, Lott JM, Hislop A, Canaday DH, Brutkiewicz RR, Blum JS. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity. 2005;22:571–581. doi: 10.1016/j.immuni.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Zwart W, Griekspoor A, Kuijl C, Marsman M, van Rheenen J, Janssen H, Calafat J, van Ham M, Janssen L, van Lith M, et al. Spatial separation of HLA-DM/HLA-DR interactions within MIIC and phagosome-induced immune escape. Immunity. 2005;22:221–233. doi: 10.1016/j.immuni.2005.01.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.