

Abstract
The phytochemical resveratrol, which is found in grapes and wine, has been reported to have a variety of anti-inflammatory, anti-platelet, and anti-carcinogenic effects. Based on its structural similarity to diethylstilbestrol, a synthetic estrogen, we examined whether resveratrol might be a phytoestrogen. At concentrations (≈3–10 μM) comparable to those required for its other biological effects, resveratrol inhibited the binding of labeled estradiol to the estrogen receptor and it activated transcription of estrogen-responsive reporter genes transfected into human breast cancer cells. This transcriptional activation was estrogen receptor-dependent, required an estrogen response element in the reporter gene, and was inhibited by specific estrogen antagonists. In some cell types (e.g., MCF-7 cells), resveratrol functioned as a superagonist (i.e., produced a greater maximal transcriptional response than estradiol) whereas in others it produced activation equal to or less than that of estradiol. Resveratrol also increased the expression of native estrogen-regulated genes, and it stimulated the proliferation of estrogen-dependent T47D breast cancer cells. We conclude that resveratrol is a phytoestrogen and that it exhibits variable degrees of estrogen receptor agonism in different test systems. The estrogenic actions of resveratrol broaden the spectrum of its biological actions and may be relevant to the reported cardiovascular benefits of drinking wine.
Resveratrol (trans-3, 4′, 5-trihydroxystilbene) occurs naturally in grapes and a variety of medicinal plants. In plants, resveratrol functions as a phytoalexin that protects against fungal infections (1). Because of its high concentration in grape skin, significant amounts of resveratrol are present in wine (2, 3), and it has been proposed to explain, at least in part, the apparent ability of moderate consumption of red wine to reduce the risk of cardiovascular disease (4–7). Resveratrol also has been reported to have cancer chemopreventive activity (8). The similarity in structure between resveratrol and the synthetic estrogen diethylstilbestrol (DES; 4, 4′-dihydroxy-trans-α, β-diethylstilbene) prompted us to investigate whether resveratrol might exhibit estrogenic activity, a property that is known to produce a cardioprotective benefit (9, 10).
Estrogens, including phytoestrogens, act via the estrogen receptor, a member of the nuclear receptor superfamily. Estrogen binding to the receptor activates the transcription of estrogen-responsive target genes. We report here that resveratrol binds to and activates transcription by the estrogen receptor at concentrations that are comparable to those required for its other biological effects.
EXPERIMENTAL PROCEDURES
Cell Culture.
MCF-7 cells, subclone WS8 (estrogen receptor-positive), MDA-MB-231 cells, subclone 10A (estrogen receptor-negative), and T47D cells, subclone A18 (estrogen receptor-positive) are derived from human breast adenocarcinomas and were provided by V. Craig Jordan (Northwestern University Medical School, Chicago). MCF-7 and MDA-MB-231 cells were grown in MEM supplemented with nonessential amino acids, 10 mM Hepes, and 5% calf serum. T47D cells were grown in RPMI 1640 medium supplemented with nonessential amino acids, 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate. BG-1 (human ovarian adenocarcinoma, estrogen receptor-positive) cells, provided by Jeff Boyd, University of Pennsylvania Medical Center, Philadelphia, were grown in DMEM/Ham’s F-12 medium with 10% fetal bovine serum. Estrogen-depleted media contained no phenol red and were supplemented with sera extracted three times with dextran-coated charcoal.
Estrogen Receptor Binding Assay.
Cytosol was prepared from MCF-7 cells grown for 4 days in estrogen-depleted medium. Cells were incubated for 30 min in 10 volumes of ice-cold hypotonic TESH buffer (10 mM Tris⋅HCl/1.5 mM EDTA/1 mM DTT, pH 7.4) (11) containing 1 mM phenylmethylsulfonyl fluoride and 10 μg/ml leupeptin and lysed by 10 passages through a 25-gauge hypodermic needle. After ultracentrifugation (1 h at 100,000 × g), the cytosol (supernatant) fraction was adjusted to 10% glycerol and stored at −70°C.
Aliquots containing 12 μg of protein were incubated with various concentrations of 16α-[125I]-iodo-3,17β-estradiol (New England Nuclear; 2200 Ci/mmol) and resveratrol or estradiol in TEGDMo buffer (40 mM Tris⋅HCl, pH 7.4/1 mM EDTA/10% glycerol/10 mM DTT/10 mM Na2MoO4, pH 7.4) overnight at 4°C in a final volume of 100 μl (12). Bound and unbound 125I-estradiol were separated with dextran-coated charcoal (11) and measured in a γ counter. Nonspecific binding was determined in the presence of a 500-fold excess unlabeled estradiol or DES and was subtracted from total binding.
Plasmids.
The plasmid tk109-luc was constructed by inserting the −109 to +52 fragment of the herpes simplex virus thymidine kinase (tk) gene into pA3luc (13, 14). Plasmids ERE-tk109-luc and ERE2-tk109-luc were constructed by cloning single or double copies of the Xenopus vitellogenin A2 estrogen response element (ERE) (15) into the HindIII site of tk109-luc. ERE-tk81-luc contains a single ERE upstream of the minimal thymidine kinase promoter (−81 to +52) in pA3luc. CMV-luc (16) and FOS-luc (17), which contain the cytomegalovirus and c-fos promoters, respectively, were used as estrogen-nonresponsive controls. The wild-type human estrogen receptor expression vector pSG5-HEGO was provided by Pierre Chambon (Université Louis Pasteur, Strasbourg, France) (18), and the pSG5 control plasmid was purchased from Stratagene.
Transfection and Luciferase Assays.
Cells were grown in estrogen-depleted media and transfected by using liposomes of dioleyl phosphatidylethanolamine and dimethyldioctadecylammonium bromide (Sigma) (19, 20). MCF-7 and MDA-MB-231 cells were transfected in 6-well plates by using 7.5 μg/well and 3 μg/well of the two lipid components, respectively, and 1 μg/well of reporter gene. BG-1 cells were transfected in 12-well plates by using 4.5 μg/well and 7.5 μg/well of the lipids and 2.5 μg/well of reporter gene. Cells were incubated with liposome-DNA complexes in serum-free, estrogen-depleted media for 6–7 h and then transferred to treatment media that contained estradiol, resveratrol, or estrogen antagonists added as stock solutions in absolute ethanol. Ethanol was added to control media to produce the same final solvent concentration (typically 0.1%) in all wells. Resveratrol, 17β-estradiol, DES, and tamoxifen were purchased from Sigma. ICI 182780 was provided by Alan Wakeling (ICI Pharmaceuticals, Macclesfield, England) and Craig Jordan (Northwestern University Medical School, Chicago). Luciferase activity (21) was determined approximately 22 h after transfection, by using an AutoLumat LB953 luminometer (EG & G, Salem, MA).
Reverse Transcription (RT)-PCR Assays for Progesterone Receptor mRNA Expression.
MCF-7 cells were grown in estrogen-depleted medium for 5 days, then treated for 24 h with ethanol (control), estradiol (0.01 or 1 nM), or resveratrol (3, 10, or 30 μM). RNA was isolated by using the RNeasy Mini Kit (Qiagen, Chatsworth, CA). Total RNA (3 μg) was subjected to RT by incubation at room temperature for 10 min followed by incubation at 42°C for 15 min by using conditions described previously (22). PCR was performed by using specific primers for the progesterone receptor gene (23) and for an internal control, glyceraldehyde-3-phosphate dehydrogenase (22). PCR reactions (50 μl) included 1 μl of the RT reaction product and 50 pmol of sense and antisense primers for the receptor and dehydrogenase genes. After 25 cycles, a 20-μl aliquot of each reaction was subjected to polyacrylamide (6%) gel electrophoresis and quantitated as described (22).
Estrogen-Dependent Cell Proliferation.
T47D cells were estrogen-depleted for 5 days and seeded into 96-well plates at 5000 cells/well. Treatment media (100 μl/well) were added on the following day and replaced at 48-h intervals until the end of the experiment. Cell density was measured via the tetrazolium reduction assay (Promega) (24) after 0, 2, 4, 6, and 8 days of culture. The absorbance (490 nM) of the formazan product was measured directly in the 96-well plates with an EL 312e microplate reader (Biotek Instruments, Luton, U.K.).
RESULTS
Resveratrol Binds to the Human Estrogen Receptor.
The structure of resveratrol is compared with DES and estradiol in Fig. 1A. The similarities are particularly striking for resveratrol and DES, which share the stilbene backbone. The phenolic A ring that is characteristic of steroidal estrogens is present in all three compounds. These structural similarities prompted us to assess whether resveratrol might interact with the estrogen receptor. Competition binding studies were performed by using extracts of MCF-7 cells, which contain large amounts of estrogen receptor. As shown in Fig. 1B, resveratrol inhibited the binding of 125I-estradiol. The degree of inhibition depended on the concentrations of both resveratrol and the labeled ligand. At 0.1 nM 125I-estradiol, the IC50 for resveratrol was ≈10 μM, indicating that it is a relatively weak ligand for the receptor. Nevertheless, this concentration is similar to those at which resveratrol exerts other biological actions (8). At higher concentrations, inhibition by resveratrol appeared to plateau. This may be because of the presence of multiple estradiol- or resveratrol-binding proteins in the MCF-7 extracts.
Figure 1.
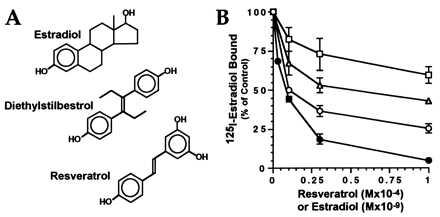
Resveratrol competes for estrogen receptor binding. (A) Structures of resveratrol, DES, and estradiol. (B) Estrogen receptor binding assays were performed as described in Experimental Procedures by using 0.1 nM (circles), 0.3 nM (triangles), or 1.0 nM (squares). 125I-estradiol competed with the indicated concentrations of resveratrol (open symbols) or unlabeled estradiol (filled circles). Each point represents the mean and range of duplicate assays after subtraction of nonspecific binding. All results are shown as percentage of binding in the absence of competitor.
Resveratrol Functions as an Agonist for Estrogen Receptor-Mediated Transcription.
The ability of resveratrol to bind to the estrogen receptor raised the possibility that it might function as an agonist or antagonist. In the presence of an agonist, the estrogen receptor initiates transcriptional activation by binding to specific EREs in the promoters of target genes (15). The actions of resveratrol were tested initially by using ERE-tk109-luc, a reporter gene that contains a single copy of an ERE upstream of the thymidine kinase promoter (Fig. 2A). In MCF-7 cells, resveratrol produced dose-dependent transcriptional activation with half-maximal induction at 5–10 μM, corresponding to the concentration required for inhibition of estradiol binding. Although considerably less potent than estradiol, resveratrol produced a maximal level of induction that was 2- to 3-fold greater than that achieved by using maximal doses of estradiol (0.1 nM). This superagonism by resveratrol was highly reproducible and was not observed with DES, which produced the same level of activation as estradiol at saturating (1 nM) concentrations (Fig. 2B). The superagonism by resveratrol also was observed with the ERE-tk81-luc reporter gene.
Figure 2.
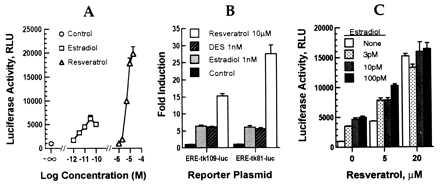
Activation of estrogen-responsive reporter plasmids by resveratrol in MCF-7 cells. (A) MCF-7 cells were transfected with ERE-tk109-luc, incubated with the indicated amounts of estradiol or resveratrol, and assayed for luciferase activity as described above. (B) Comparisons of estradiol, resveratrol, and DES using different reporter genes. (C) Effect of combined estradiol and resveratrol in the activation of ERE-tk109-luc. Results are shown as mean ± SEM for triplicate transfections.
Transfected MCF-7 cells were treated with combinations of resveratrol and estradiol to determine whether their actions were additive, synergistic, or antagonistic. Suboptimal doses of estradiol and resveratrol were additive (Fig. 2C), but maximal activation by resveratrol was not increased further by estradiol. These results are consistent with both compounds activating the same receptor.
Several additional types of control experiments were performed to confirm that the effects of resveratrol are estrogen receptor-mediated (Fig. 3). Deletion of the ERE from ERE-tk109-luc eliminated transcriptional activation by resveratrol (and estradiol) at each dose tested (Fig. 3A). This result, in combination with experiments using ERE-tk81-luc, confirms that the response of the reporter gene to resveratrol is not caused by estrogen receptor-independent activation of the basal promoter. Resveratrol did not activate other control reporter plasmids such as CMV-luc and FOS-luc, which are not estrogen-responsive (data not shown). Two estrogen antagonists, tamoxifen and ICI 182780, inhibited reporter gene activation by resveratrol (Fig. 3B). An estrogen receptor-negative cell line, MDA-MB-231, was used to confirm that estrogen receptor was required for resveratrol action (Fig. 3C). In the absence of cotransfected estrogen receptor, resveratrol was inactive whereas coexpression of the estrogen receptor with the ERE2-tk109-luc reporter gene conferred responsiveness to resveratrol. Thus, resveratrol action requires an ERE in the reporter gene and the participation of estrogen receptor, and it is inhibited by specific estrogen antagonists.
Figure 3.
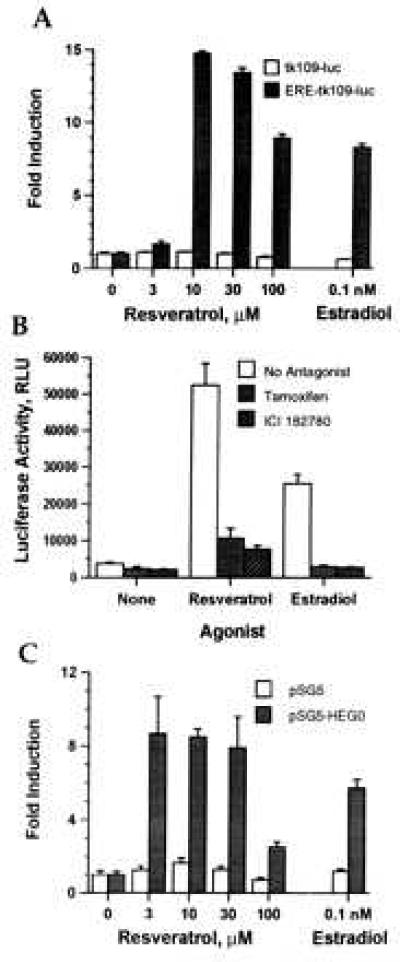
Transcriptional activation by resveratrol is estrogen receptor-mediated. (A) Dependence on an ERE. MCF-7 cells were transfected with tk109-luc or ERE-tk109-luc, incubated with the indicated concentrations of resveratrol or estradiol, and assayed for luciferase activity. Results are shown as fold induction compared with control-treated cells. (B) Inhibition by estrogen antagonists. MCF-7 cells were transfected with ERE-tk109-luc, incubated with no agonist, estradiol (0.1 nM), or resveratrol (10 μM) or with the agonists plus the antagonists tamoxifen (1 μM) or ICI 182780 (100 nM). (C) Requirement for the estrogen receptor. MDA-MB-231 cells were transfected with 1 μg/well ERE2-tk109-luc and 10 ng/well of pSG5 or pSG5-HEGO, incubated with the indicated concentrations of resveratrol or estradiol, and assayed for luciferase activity. All values are means ± SEM of triplicate transfections.
Cell-Specific Effects of Resveratrol.
The effects of resveratrol on estrogen receptor-mediated transcriptional activation of ERE2-tk109-luc were compared in human MCF-7 breast cancer cells and human BG-1 ovarian carcinoma cells, each of which contain endogenous estrogen receptors (Fig. 4). As with the single-ERE reporters, resveratrol was superagonistic in MCF-7 cells (56-fold induction vs. 24-fold by estradiol). In contrast, in BG-1 cells, transcriptional activation by resveratrol (8-fold) was somewhat less than that by estradiol (12-fold).
Figure 4.
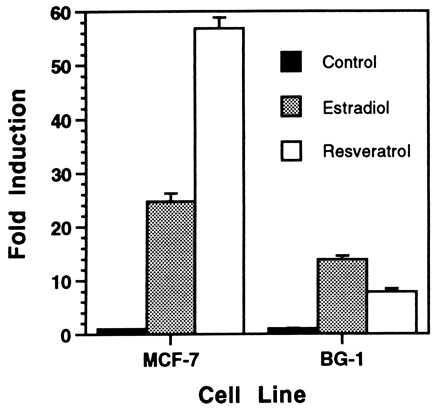
Agonistic activity of resveratrol is cell type-dependent. MCF-7 and BG-1 cells were transfected with ERE2-tk109-luc, incubated with maximally activating concentrations of estradiol and resveratrol (determined separately for each cell type), and assayed for luciferase activity. Estradiol and resveratrol concentrations were 10 pM and 10 μM, respectively, for MCF-7 cells and 1 nM and 33 μM for BG-1 cells. Results are mean ± SEM for three (MCF-7) or four (BG-1) replicate transfections.
Resveratrol Stimulates Expression of Endogenous Estrogen-Regulated Genes.
The effects of resveratrol and estradiol on expression of progesterone receptor mRNA were analyzed in MCF-7 cells to determine whether resveratrol stimulates expression of natural estrogen-regulated genes as well as transfected reporter genes. As shown in Fig. 5A, resveratrol (10–30 μM) was as effective as a maximal (1 nM) dose of estradiol in activating progesterone receptor gene expression. Resveratrol and estradiol also induced expression of the pS2 gene in MCF-7 cells, and both of these estrogen-responsive genes were stimulated in T47D cells (data not shown).
Figure 5.
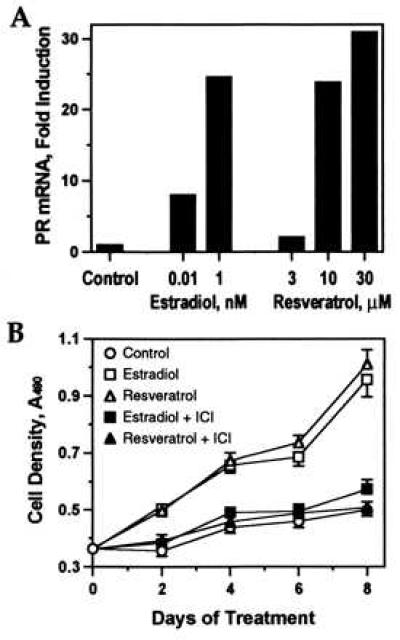
Effect of resveratrol on estrogen-regulated transcription and growth. (A) MCF-7 cells were treated with resveratrol or estradiol, and progesterone receptor (PR) mRNA was measured by RT-PCR. Results are normalized to glyceraldehyde-3-phosphate dehydrogenase mRNA and shown as fold induction relative to control (0.1% ethanol). Similar results were obtained in an independent experiment. (B) T47D cells were cultured in the presence of 0.1 nM estradiol or 10 μM resveratrol with or without 100 nM ICI 182780 or in control medium. Cell density was determined at the indicated intervals. Each point represents the mean ± SEM of eight replicate wells (n = 16 for day 0).
Resveratrol Stimulates the Proliferation of Estrogen-Dependent Breast Cancer Cells.
T47D cells, an estrogen-dependent breast cancer cell line, were used to compare the effects of resveratrol and estradiol on cell growth. As shown in Fig. 5B, resveratrol (10 μM) induced T47D proliferation to the same extent as estradiol (0.1 nM). Cells grew equally well in 0.1 or 1 nM estradiol (data not shown), indicating that this rate of growth represents maximal estrogenic stimulation. The effect of resveratrol, like that of estradiol, was blocked by the estrogen antagonist, ICI 182780.
DISCUSSION
By using several different assay systems, these studies demonstrate that resveratrol is a phytoestrogen. It competes with 125I-labeled estradiol for binding to the human estrogen receptor. Furthermore, it activates the expression of estrogen-responsive reporter genes in several different human cell lines, and this activation is inhibited by estrogen antagonists. Resveratrol also fully activates expression of endogenous estrogen-regulated genes, and it induces the proliferation of T47D breast cancer cells. The EC50 for estrogenic stimulation by resveratrol ranged between 3 and 10 μM, depending on the test system examined. Thus, the estrogenic effects of resveratrol occur at concentrations that are similar to those required for its reported anti-inflammatory (25, 26), anti-platelet (6), and anti-carcinogenic (8) activities.
Resveratrol produced greater maximal transcriptional activation than estradiol, but this superagonism was not seen in all cell types. For example, resveratrol produced two to four times greater activation of reporter plasmids than estradiol in MCF-7 breast cancer cells but less activation than estradiol in BG-1 ovarian carcinoma cells. These cell type-specific effects of resveratrol are reminiscent of the well known tissue-specific and species-specific effects of agents such as tamoxifen (27), which can act as an estrogen receptor agonist in some tissues such as the uterus but acts as an estrogen antagonist in the breast (27). Other recently characterized estrogen receptor ligands such as raloxifene also appear to exert tissue-selective actions (28). Superagonism was not observed in the induction of progesterone receptor expression, even in MCF-7 cells. Although this result may reflect differences in endogenous vs. transfected genes, it suggests that the effects of resveratrol may vary depending on the target gene as well as the cell type.
The mechanisms for gene- and tissue-specific effects of estrogen receptor ligands are not known. It has been shown recently that a second form of estrogen receptor exists (estrogen receptor β) (29). The different tissue distributions of the estrogen receptors α and β might contribute to the differential effects of various estrogen receptor ligands, including resveratrol. In addition, expression of different transcriptional coactivators and corepressors in various tissues (reviewed in ref. 30) and the ability of different ligands to induce various conformations of the estrogen receptor (31) may result in tissue- and ligand-specific activation of certain genes. It is also possible that, in some cell types (e.g., MCF-7), resveratrol produces superagonism by activating additional signaling pathways that converge on estrogen receptor-mediated transcription.
The potential biological impact of environmental and dietary estrogens on human health has generated considerable interest (32–34). These agents include phytoestrogens as well a variety of synthetic compounds. Chemically, many of the known phytoestrogens are flavonoids; others are coumestans or resorcylic acid lactones (35, 36), and an estrogenic hydroxystilbene recently has been reported to occur naturally in wood (37). The finding that resveratrol is estrogenic expands the spectrum of known dietary phytoestrogens.
Red wine appears to be more estrogenic than bourbon or beer, which contain other phytoestrogens but not resveratrol (38). Resveratrol is found primarily in the skin of grapes and is therefore relatively abundant in red, but not white, wines. There is considerable variation in the resveratrol content even among red wines, depending on grape cultivar, vintage, and place of origin and also on the analytical technique used for measurement. Concentrations of 10–20 μM are common although higher or lower values also are found frequently (3). In addition to free resveratrol, wines contain resveratrol glycosides that may contribute to the biologically available dose (39). Unfortunately, no data have been published concerning serum levels of resveratrol after wine consumption, and there is no information about its rate of clearance from the bloodstream, the identities and activities of its metabolic products, or the potential first-pass effects of portal circulation through the liver. In the absence of such data, the physiological significance of resveratrol in wine remains uncertain.
However, the anti-oxidant (4) and anti-platelet (6, 40) activities of resveratrol have been invoked as possible mechanisms for the reported cardiovascular benefits of moderate wine consumption and the so-called “French paradox” (7). Goldberg and coworkers (6) found that resveratrol inhibited thromboxane B2 synthesis and thrombin-induced aggregation of human platelets in vitro, with IC50s of 7 μM and ≈160 μM, respectively. More recently, they examined the effects of dietary supplementation with resveratrol and found that platelets from human volunteers who consumed 2 mg (≈9 μmol) per day showed diminished thromboxane B2 synthesis and reduced thrombin-induced aggregation compared with controls (41). A few glasses of many red wines could supply this amount of resveratrol. These results suggest that daily consumption of some red wines might produce pharmacologically significant concentrations of resveratrol in the blood. Based on the concentrations required in vitro, it seems likely that doses of resveratrol that affect platelet behavior could also have some estrogenic effect. It is intriguing to consider whether the estrogenicity of resveratrol may contribute to the reported cardiovascular benefits of red wine (7). Estrogens have similar effects if administered orally; the high doses delivered to the liver via portal circulation produce beneficial changes in serum lipids (9, 10). Similarly, wine consumption may expose the liver to higher concentrations of resveratrol than occur in the systemic circulation.
The finding that resveratrol stimulates the growth of human breast cancer cells is also potentially significant. Although Jang et al. (8) found that resveratrol exerts an anticarcinogenic effect in mouse mammary cultures, our results suggest that resveratrol could exert a growth-stimulating estrogenic effect on human breast carcinomas. This apparent contradiction might be explained by the observation that, although many human breast cancers are mitogenically stimulated by estrogen, most mouse mammary cancers are estrogen-insensitive (42). Thus, although the anti-carcinogenic and anti-thrombotic activities of resveratrol show pharmacological promise, its estrogenic properties may produce undesirable side effects and limit the circumstances under which it can be used safely.
Further studies are required to assess the physiological significance of resveratrol in humans, and a more complete understanding of its estrogenic actions is needed to understand its role as a dietary substance. The superagonistic properties of resveratrol raise the possibility that structure–function studies could lead to the development of more selective estrogen receptor agonists and antagonists, which could be useful as a therapeutic agents.
Acknowledgments
We thank V. Craig Jordan for the generous gifts of cells, advice, and reagents, Laird Madison for assistance with the receptor binding assays, Teresa Woodruff for assistance with the microplate reader, and Wade Johnson for helpful discussions and advice. We are grateful to Pierre Chambon, Takashi Nagaya, Richard Pestell, and Srividya Sundaresan for plasmids, Jeff Boyd for cell lines, and Alan Wakeling for ICI 182780. This work was funded by the U.S. Army Medical Research and Materiel Command Breast Cancer Research Program Grants DAMD17-94-J-4082 (to J.L.J.) and DAMD17-96-1-6018 (to J.M.M.).
ABBREVIATIONS
- DES
diethylstilbestrol
- ERE
estrogen response element
- RT
reverse transcription
References
- 1.Hain R, Bieseler B, Kindl H, Schroder G, Stocker R. Plant Mol Biol. 1990;15:325–335. doi: 10.1007/BF00036918. [DOI] [PubMed] [Google Scholar]
- 2.Siemann E H, Creasy L L. Am J Enol Vitic. 1992;43:49–52. [Google Scholar]
- 3.Goldberg D M, Karumanchiri A, Yan J, Soleas G, Ng E, Waterhouse A L, Diamandis E P. Am J Enol Vitic. 1995;46:159–165. [Google Scholar]
- 4.Frankel E N, Waterhouse A L, Kinsella J E. Lancet. 1993;341:1103–1104. doi: 10.1016/0140-6736(93)92472-6. [DOI] [PubMed] [Google Scholar]
- 5.Bertelli A A, Giovannini L, Giannessi D, Migliori M, Bernini W, Fregoni M, Bertelli A. Int J Tissue React. 1995;17:1–3. [PubMed] [Google Scholar]
- 6.Pace-Asciak C R, Hahn S, Diamandis E P, Soleas G, Goldberg D M. Clin Chim Acta. 1995;235:207–219. doi: 10.1016/0009-8981(95)06045-1. [DOI] [PubMed] [Google Scholar]
- 7.Goldberg D M, Hahn S E, Parkes J G. Clin Chim Acta. 1995;237:155–187. doi: 10.1016/0009-8981(95)06069-p. [DOI] [PubMed] [Google Scholar]
- 8.Jang M, Cai L, Udeani G O, Slowing K V, Thomas C F, Beecher C W, Fong H H, Farnsworth N R, Kinghorn A D, Mehta R G, Moon R C, Pezzuto J M. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 9.Lobo R A. Am J Obstet Gynecol. 1995;173:982–989. doi: 10.1016/0002-9378(95)90247-3. [DOI] [PubMed] [Google Scholar]
- 10.Odell W D. In: Endocrinology. 3rd ed. DeGroot L J, Besser M, Burger H G, Jameson J L, Loriaux D L, Marshall J C, Odell W D, Potts J T Jr, Rubenstein A H, editors. Philadelphia: Saunders; 1995. pp. 2128–2139. [Google Scholar]
- 11.Clark J H, Peck J, Jr, E J, Markaverich B M. In: Laboratory Methods: Manual for Hormone Action and Molecular Endocrinology. Schrader W T, O’Malley B W, editors. Baylor College of Medicine, Houston, TX: Department of Cell Biology; 1987. [Google Scholar]
- 12.Kuiper G G J M, Carlsson B, Grandien K, Enmark E, Häggblad J, Nilsson S, Gustafsson J-Å. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 13.Wood W M, Kao M Y, Gordon D F, Ridgway E C. J Biol Chem. 1989;264:14840–14847. [PubMed] [Google Scholar]
- 14.Nagaya T, Madison L D, Jameson J L. J Biol Chem. 1992;267:13014–13019. [PubMed] [Google Scholar]
- 15.Klein-Hitpass L, Schorpp M, Wagner U, Ryffel G U. Cell. 1986;46:1053–1061. doi: 10.1016/0092-8674(86)90705-1. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe G, Howe A, Lee R J, Shu I-W, Albanese C, Karnezis A, Kyriakis J, Zon L, Rundell K, Pestell R G. Proc Natl Acad Sci USA. 1996;93:12861–12866. doi: 10.1073/pnas.93.23.12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sundaresan S, Colin I M, Pestell R G, Jameson J L. Endocrinology. 1996;137:304–311. doi: 10.1210/endo.137.1.8536629. [DOI] [PubMed] [Google Scholar]
- 18.Tora L, Mullick A, Metzger D, Ponglikitmongkol M, Park I, Chambon P. EMBO J. 1989;8:1981–1986. doi: 10.1002/j.1460-2075.1989.tb03604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rose J K, Buonocore L, Whitt M A. Biotechniques. 1991;10:520–525. [PubMed] [Google Scholar]
- 20.Campbell M J. BioTechniques. 1995;18:1027–1032. [PubMed] [Google Scholar]
- 21.deWet J R, Wood K V, DeLuca M, Helinski D R, Subramani S. Mol Cell Biol. 1987;7:725–737. doi: 10.1128/mcb.7.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson W, Albanese C, Handwerger S, Williams T, Pestell R G, Jameson J L. J Biol Chem. 1997;272:15405–15412. doi: 10.1074/jbc.272.24.15405. [DOI] [PubMed] [Google Scholar]
- 23.Tang P-Z, Gannon M J, Andrew A, Miller D. Eur J Endocrinol. 1995;133:598–605. doi: 10.1530/eje.0.1330598. [DOI] [PubMed] [Google Scholar]
- 24.Cory A H, Owen T C, Barltrop J A, Cory J G. Cancer Comm. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 25.Kimura Y, Okuda H, Arichi S. Biochim Biophys Acta. 1985;834:275–278. [PubMed] [Google Scholar]
- 26.Kimura Y, Okuda H, Kubo M. J Ethnopharmacol. 1995;45:131–139. doi: 10.1016/0378-8741(94)01206-f. [DOI] [PubMed] [Google Scholar]
- 27.Jordan V C, Murphy C S. Endocr Rev. 1990;11:578–610. doi: 10.1210/edrv-11-4-578. [DOI] [PubMed] [Google Scholar]
- 28.Sato M, Rippy M K, Bryant H U. FASEB J. 1996;10:905–912. doi: 10.1096/fasebj.10.8.8666168. [DOI] [PubMed] [Google Scholar]
- 29.Kuiper G G, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson J A. Proc Natl Acad Sci USA. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horwitz K B, Jackson T A, Bain D L, Richer J K, Takimoto G S, Tung L. Mol Endocrinol. 1996;10:1167–1177. doi: 10.1210/mend.10.10.9121485. [DOI] [PubMed] [Google Scholar]
- 31.Beekman J M, Allan G F, Tsai S Y, Tsai M J, O’Malley B W. Mol Endocrinol. 1993;7:1266–1274. doi: 10.1210/mend.7.10.8264659. [DOI] [PubMed] [Google Scholar]
- 32.Cotton P. J Am Med Assoc. 1994;271:414–416. doi: 10.1001/jama.271.6.414. [DOI] [PubMed] [Google Scholar]
- 33.Safe S H. Environ Health Perspect. 1995;103:346–351. doi: 10.1289/ehp.95103346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feldman D. Endocrinology. 1997;138:1777–1779. doi: 10.1210/endo.138.5.5213. [DOI] [PubMed] [Google Scholar]
- 35.Miksicek R J. Mol Pharmacol. 1993;44:37–43. [PubMed] [Google Scholar]
- 36.Miksicek R J. J Steroid Biochem Molec Biol. 1994;49:153–160. doi: 10.1016/0960-0760(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 37.Mellanen P, Petänen T, Lehtimäki J, Mäkelä S, Bylund G, Holmbom B, Mannila E, Oikari A, Santti R. Toxicol Appl Pharmacol. 1996;136:381–388. doi: 10.1006/taap.1996.0046. [DOI] [PubMed] [Google Scholar]
- 38.Gavaler J S, Rosenblum E R, Deal S R, Bowie B T. Proc Soc Exp Biol Med. 1995;208:98–102. doi: 10.3181/00379727-208-43839. [DOI] [PubMed] [Google Scholar]
- 39.Goldberg D M, Diamandis E P, Ng E, Soleas G, Karumanchiri A. Am J Enol Vitic. 1996;47:415–420. [Google Scholar]
- 40.Bertelli A A, Giovannini L, Bernini W, Migliori M, Fregoni M, Bavaresco L, Bertelli A. Drugs Exp Clin Res. 1996;22:61–63. [PubMed] [Google Scholar]
- 41.Pace-Asciak C R, Rounova O, Hahn S E, Diamandis E P, Goldberg D M. Clin Chim Acta. 1996;246:163–182. doi: 10.1016/0009-8981(96)06236-5. [DOI] [PubMed] [Google Scholar]
- 42.Nandi S, Guzman R C, Yang J. Proc Natl Acad Sci USA. 1995;92:3650–3657. doi: 10.1073/pnas.92.9.3650. [DOI] [PMC free article] [PubMed] [Google Scholar]