Figure 18.1 Normal dural investment of the superior sagittal dural venous sinus. A, Coronal graphic demonstrates the normal splitting of the periosteal/endosteal (outer dura) and (inner) meningeal dura (black arrows) to encompass the superior sagittal dural venous sinus. Also note the penetration of a bridging vein through the dural wall of the venous sinus (white arrow). B, Coronal T2WI MRI following craniotomy and corpus callosotomy demonstrates hyperintense extradural postoperative fluid abutting the periosteal dura (arrowhead). Graphic used with permission from Amirsys, Inc., Osborn’s Brain: Imaging. Pathology and Anatomy, 2012.
Dural border cell layer
Moving inward, the transition between the meningeal dura and the innermost dura is characterized by decreasing collagen fibers and an increase in amorphous extracellular material (8, 13). The term DBCL describes the inner layer of the dura that is in intimate contact with the arachnoid. The composition of this inner DBCL is unique in three ways. First, unlike the meningeal and endosteal dura, extracellular collagen is lacking or sparse in the DBCL. Secondly, the spaces between the sparsely arrayed dural border cells are varied in size. Finally, there are relatively few tight cell junctions between the dural border cells. These three characteristics of the normal DBCL create a structurally weak cellular interface at the DBCL–arachnoid junction (8, 13–15). Adeeb et al. and Croft and Reichard have observed that the DBCL is directly applied to the arachnoid mater (arachnoid barrier cells) with no or few recognizable junctions between them (4, 8). Clinical neuropathologic observations that support a tight relationship between the arachnoid barrier cells and DBCL layer include: (1) SDHs are not always associated with subarachnoid hemorrhage (SAH) (unless there is tearing of arachnoid vessels including cortical dural bridging veins); (2) purulent meningitis remains confined to the SAS and rarely is associated with subdural empyema; (3) SAS hemorrhage, as might be seen with ruptured aneurysm, does not typically extravasate into the subdural compartment (pers. comm. with Lucy B. Rorke-Adams MD).
Arachnoid mater
The arachnoid mater and specifically the arachnoid barrier cells are in contact with the inner dural border cells. There are no or few recognizable junctions between them. The separation between these two cellular planes creates what is commonly known as the subdural space or using the language of Orlin, “the subdural compartment” (14). The arachnoid mater and the pia mater are normally interconnected by a honeycomb network of trabeculae. Cerebrospinal fluid (CSF) fills these interstices, thus creating the SAS. It is important to recognize that pathologic processes (e.g., hemorrhage, infection, and tumor) that originate within the SAS tend to remain within this compartment (2, 16).
Pia mater
The pia mater is a delicate yet adherent vascular fibroelastic membrane closely investing the brain and spinal cord. It hugs the cerebral convolutions, passing as a double layer into the cerebral hemispheric sulci and folial fissures of the cerebellum. Folding back upon itself, it forms the tela choroidea of the third and fourth ventricles and invaginates into the ventricles to form the choroid plexus (9).
Summary
The accumulation of hemorrhage in and adjacent to these meningeal boundaries and the adjacent anatomic compartments, either normally pre-existing or pathologically created, demonstrates characteristic morphologic appearances and thus are of particular importance to the radiologist and neuropathologist.
In summary, between the arachnoid and pia, a normal SAS exists with normal more focally prominent intracranial SASs called intracranial cisterns. There is no naturally existing sudural space. Blood or nonhemorrhagic fluid in the subdural compartment reflects a pathologic state of either traumatic or nontraumatic origin. At the time of autopsy, a subdural compartment may be artifactually created. The pia tightly hugs the cortical gray matter and focal hemorrhage here is difficult to distinguish from peripheral parenchymal hemorrhage. Subpial hemorrhage does not conform to a cerebral sulcus and will lack change with patient re-positioning, thus distinguishing it from SAH.
Hemorrhagic and nonhemorrhagic extra-axial collections: the foundations
As an introduction to this section and to serve as a foundation for subsequent sections, a focused discussion of the computed tomography (CT) and magnetic resonance imaging (MRI) characteristics of intracranial extra-axial hemorrhage will follow.
In the setting of pediatric head trauma, including AHT and AccT, the detection of intracranial extra-axial hemorrhage is common, can be observed in one or more compartments, and may be associated with underlying brain injury (6, 7, 16–24). The possible locations for intracranial nonparenchymal hemorrhage include the epidural space, the SAS, the subdural compartment, the ventricles, and the subpial compartment (subpial hemorrhage). Although these hemorrhages may exist in the absence of associated brain injury, their presence should always alert the radiologist to the possibility that extra-axial hemorrhage may represent a proxy or marker for serious injury of the underlying brain (see Chapter 19) (25–30). The natural history and prognosis differ for hemorrhage in these various compartments. This is particularly true of EDH and SDH (31, 32). The rapid detection and characterization of intracranial hemorrhage leads to accurate triage of those patients who require life-saving surgical decompression and those who warrant nonsurgical management with serial imaging to fully characterize the magnitude of injury (33).
A multidisciplinary working understanding, clear usage, and acceptance of terms that describe hemorrhagic and nonhemic fluid in the subdural compartment (mixed attenuation SDH, hematohygroma, hygroma, cSDH, subdural effusion, and subdural empyema) facilitates clinical care, professional communication, and clarifies medico-legal proceedings (16, 22).
Estimating the age of extra-axial hemorrhage using CT and MRI
In the process of clinical assessment, CPS investigation, and judicial inquiry, establishing whether trauma was perpetrated is paramount. Additionally, the understanding of injury type (e.g., AHT vs. AccT), and estimation of the age of injury including extra-axial hemorrhage are critical pieces of information. The important contribution of accurate neuroimaging information added to all gathered data cannot be overstated and influences the consideration of potential suspects in the setting of AHT.
In the setting of suspected AHT, there may be no relevant history or a changing history is provided. When a history of trauma is offered, the purported mechanism may not match the magnitude of assessed ICI, or imaging evidence may indicate injuries of differing ages. Thus, estimating the age of extra-axial hemorrhage with imaging technologies (e.g., CT and MRI) provides critical forensic information for the investigation of suspected AHT (7, 34, 35).
For the radiologist, the common clinical questions are: What is the age of the SDH(s); are there differing ages of hemorrhage; and is there injury to the brain? CT and MRI are complementary imaging tools and indispensable for tackling the challenge of characterizing and dating intracranial hemorrhage, particularly SDH (20, 26). Serial imaging plays an important role in clarifying the age of mixed attenuation noncontrast-enhanced CT (NECT), and/or heterogeneous signal intensity (MRI) subdural compartment hemorrhage (33, 36, 37).
For the radiologist, pinpointing the precise age of extra-axial hemorrhage is fraught with pitfalls and frankly, is unrealistic. It cannot be overemphasized that at the time of initial imaging of the child with extra-axial hemorrhage (usually NECT), and in the few days following injury, the radiologist provides the most useful support by clearly describing the attenuation (NECT) and/or signal intensity (MRI) features of the hemorrhage rather than attempting to date the age of the hemorrhage. To emphasize the potential pitfall of early hemorrhage dating with NECT, consider the child who presents with an iso-to-hypoattenuating SDH on NECT. The hypoattenuating component(s) could represent hyperacute, late subacute (2–3 weeks of age), or acute hemorrhage in an anemic child or CSF (hygroma) (Table 18.1).
| Time | Stage | CT attenuation |
|---|---|---|
| Less than 3 hours | Hyperacute | Hypo-to-isoattenuating* |
| 3 hours to 3 days | Acute | Hyper- or mixed hyper–hypo |
| 3 days to 7–10 days | Early subacute | Hyperattenuating |
| 7–10 days to 3 weeks | Late subacute | Isoattenuating* |
| >3 weeks | Chronic | Hypoattenuating |
* SDH is isoattenuating when hemoglobin levels are <8–10% or when hyperacute.
Serial imaging with CT and/or MRI, although often underutilized, proves invaluable in heightening the accuracy of estimating hemorrhage age (23, 24, 33, 36, 37). Resisting the tendency to immediately default to a radiologic impression that reflects precise dating language in the imaging report will not only improve the radiologist’s reporting accuracy but will best support the multidisciplinary collaboration of the Child Protection Team (CPT).
There are imaging findings (CT and MRI) that strongly suggest an acute or early subacute ICI. These include: scalp and subscalp edema, cerebral edema and/or ischemia, and imaging evidence of recent (hyperacute, acute, or subacute) extra-axial hemorrhage (i.e., the absence of scalp edema does not exclude hyperacute, acute, or early subacute injury). Remote injury is suggested by extra-axial hemorrhage with NECT or MRI features consistent with late subacute or chronic hemorrhage, including a large size of the extra-axial hemorrhage, the presence of membrane structure within the SDH, the lack of edema or ischemia, and evidence of underlying brain atrophy or encephalomalacia (22, 38, 39).
NECT characterization of extra-axial hemorrhage
There are many factors that influence the imaging appearance of extra-axial hemorrhage. The NECT attenuation of hemorrhage is by convention described in comparison to the attenuation of the brain parenchyma and reported in Hounsfield units (HU) (25, 26, 40). The concentration of hemoglobin, compactness of clot retraction, and presence of serum protein influence the appearance of hemorrhage on NECT (41). Interestingly, it is the globin or protein that contributes primarily to blood’s attenuation characteristics. The iron within hemoglobin contributes only 8–10% of the overall attenuation (38, 40).
Within the first few hours (<3 h), extra-axial hemorrhage is considered hyperacute, and will appear iso-to-hypoattenuating relative to normal brain parenchyma. Beyond this early hyperacute period and for the remainder of the first three post-trauma days, clot retraction and hemoglobin concentration lead to an increase in attenuation of the hyperattenuating clot. Factors that may lead to an isoattenuating rather than hyperattenuating acute SDH (aSDH) include the admixture of acute hemorrhage with hyperacute blood or CSF (hematohygroma). Acute SDH in a child who is anemic (e.g., hemoglobin values <8–10 g/dL) may also be isoattenuating. In profound states of anemia, the SDH may be hypoattenuating (22). The evolution of hemorrhage aging within the subdural compartment is that of progressive decreasing attenuation as a result of clot proteolysis. This evolves at a rate of approximately 1.5 HU per day (Fig. 18.2). Thus, by days 7–10 the SDH has become isoattenuating to cerebral gray matter and after 2–3 weeks is hypoattenuating (see Table 18.1). Of course, interval bleeding due to an active bleeding source or coagulopathy, CSF admixture, or anemia will alter the attenuation values, and thus make dating the age of hemorrhage more challenging (16, 26–28).
 Figure 18.2 Temporal evolution of CT attenuation. The SDH will decrease in attenuation by approximately 1.5 HU/day. Note that at 7–10 days the hemorrhage is isoattenuating to cerebral cortex and by 10 days is hypoattenuating. Used with permission from Amirsys, Inc., Osborn’s Brain: Imaging. Pathology and Anatomy, 2012.
Figure 18.2 Temporal evolution of CT attenuation. The SDH will decrease in attenuation by approximately 1.5 HU/day. Note that at 7–10 days the hemorrhage is isoattenuating to cerebral cortex and by 10 days is hypoattenuating. Used with permission from Amirsys, Inc., Osborn’s Brain: Imaging. Pathology and Anatomy, 2012.The NECT imaging observation of mixed attenuation traumatic subdural compartment hemorrhage has historically invoked the default concern over acute on chronic hemorrhage (7, 42, 43). However, in addition to hemorrhage of differing ages, there are three other important differential diagnostic considerations that the radiologist must entertain. These include: (1) hyperacute and acute hemorrhage; (2) acute hemorrhage alone (e.g., clot and serum separation contribute to mixed attenuation) (Fig. 18.3); and (3) acute hemorrhage mixed with CSF (hematohygroma) (26, 44, 45). These three causes of mixed attenuation SDH often result from a single traumatic event and are often associated with ipsilateral cerebral hemispheric edema (see the Subdural hemorrhage section for further discussion). Chronic SDH on NECT is hypoattenuating to normal brain parenchyma but slightly hyperattenuating to normal CSF (e.g., sulci, cisterns, and ventricles) (Fig. 18.4).
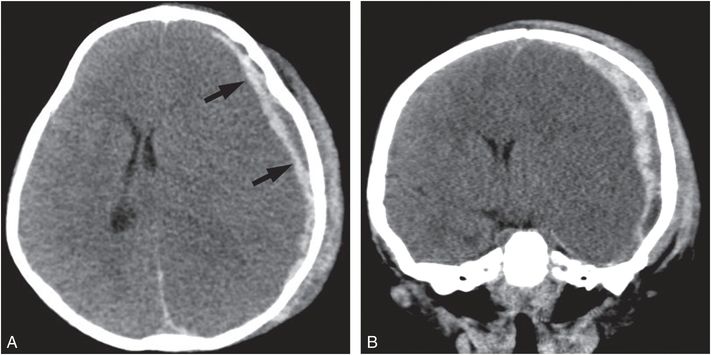 Figure 18.3 Mixed attenuation aSDH. A, B, Axial and coronal (reformatted) NECT of a seven-month-old infant with acute onset of seizure and obtundation. The left supratentorial mixed attenuation SDH (arrows), was diagnosed as acute by the pediatric neurosurgeon at the time of emergent craniectomy and hematoma evacuation. A torn, bleeding left vein of Labbe was identified. Note the associated left subgaleal hematoma, diffuse left cerebral hemispheric edema, and resultant subfalcine herniation. The assailant admitted to shaking and impacting the infant’s head against a crib.
Figure 18.3 Mixed attenuation aSDH. A, B, Axial and coronal (reformatted) NECT of a seven-month-old infant with acute onset of seizure and obtundation. The left supratentorial mixed attenuation SDH (arrows), was diagnosed as acute by the pediatric neurosurgeon at the time of emergent craniectomy and hematoma evacuation. A torn, bleeding left vein of Labbe was identified. Note the associated left subgaleal hematoma, diffuse left cerebral hemispheric edema, and resultant subfalcine herniation. The assailant admitted to shaking and impacting the infant’s head against a crib.  Figure 18.4 Chronic SDH. A six-month-old child with skeletal findings compatible with inflicted trauma. A, Axial graphic shows a left holocranial chronic SDH. B, Axial NECT depicts bilateral frontotemporal chronic SDHs of higher attenuation than CSF. Graphic used with permission from Amirsys, Inc., Osborn’s Brain: Imaging. Pathology and Anatomy, 2012.
Figure 18.4 Chronic SDH. A six-month-old child with skeletal findings compatible with inflicted trauma. A, Axial graphic shows a left holocranial chronic SDH. B, Axial NECT depicts bilateral frontotemporal chronic SDHs of higher attenuation than CSF. Graphic used with permission from Amirsys, Inc., Osborn’s Brain: Imaging. Pathology and Anatomy, 2012.MRI characterization of extra-axial hemorrhage
Our knowledge of the MRI signal intensity of extra-axial hemorrhage has come first from the study of intracranial intra-axial (i.e., parenchymal) hemorrhage (46–50). More recently, Duhem et al. reported the temporal MRI changes of SDH studied at 1.5 Tesla (as shown in Table 18.2) (36). Radiologists must recognize that the imaging appearance of SDH in the neonate, infant, and young child follows a time course that cannot be precisely extrapolated from data that has been derived from MRI observations of intracerebral hematoma. This re-emphasizes the importance of providing a dating range of hemorrhage age rather than attempting to precisely date. Additionally, the added value of complementary NECT and serial brain imaging cannot be overstated.
| Acute (Hours—3 days) | Early subacute (3—10 days) | Late subacute (10 days—3 weeks) | Chronic (over 3 weeks) | |
|---|---|---|---|---|
| Subdural hemorrhage | ||||
| T1 | Isointense |  |
 |
 |
| T2 |  |
Iso –to- | ||
| Hemoglobin | Deoxyhemoglobin | Methemoglobin(intracellular) | Methemoglobin(extracellular) | Hemosiderin |
| Intracerebral hemorrhage | ||||
| T1 | Iso – to-  |
 |
 |
Isointense |
| T2 |  | |||
| Hemoglobin | Deoxyhemoglobin | Methemoglobin(intracellular) | Methemoglobin(extracellular) | Hemosiderin |
Interpreting MR signal intensity features of hemorrhage within the extra-axial compartment is complex and influenced by several physical and technical factors (51–53). Physical factors include the concentration of tissue thromboplastin and local oxygen concentration that varies between the intra-axial and extra-axial compartments, which influence the MRI characterization of hemorrhage. Other factors that contribute to the complex MRI appearance of extra-axial hemorrhage include red blood cell(RBC) hydration, cell compactness, and cell membrane integrity. The oxidation state of hemoglobin is also an important factor. The initial oxidation step is to deoxyhemoglobin, followed in the early and late subacute periods by further oxidation to methemoglobin (Table 18.2). During the transition between the early and late subacute stages, RBC cell membrane breakdown occurs and is associated with the extracellular accumulation of methemoglobin. In this oxidation process the iron ion acquires paramagnetic properties while changing from the ferrous to the ferric state. After approximately three weeks, hemosiderin predominates (as shown in Table 18.2). To make matters of MRI hemorrhage interpretation a bit more complex, MRI technical factors such as magnetic field strength (BO) and pulse sequence selection influence the appearance of hemorrhage (50, 54, 55).
As with NECT, there is a general acceptance of the importance of establishing a dating range when reporting the MR appearance of extra-axial and intraparenchymal hemorrhage. Duhem et al., reporting their observations of intracranial SDH characterization performed at 1.5 Tesla, demonstrated acute hemorrhage (e.g., several hours to 3 days) showing isointensity to brain parenchyma on T1 weighted image (WI) and hypointensity on T2WI. Early subacute hemorrhage (e.g., 3–10 days) when clot retraction is maximal and RBC membranes are predominantly intact showed hyperintensity on T1WI and iso-to-hyperintensity on T2WI. Near the end of the first week, the RBCs within the SDH begin to lyse and the extracellular methemoglobin concentration increases. In this late subacute stage (10 days to <3 weeks) the SDH demonstrates hyperintensity on both T1WI and T2WI. In the chronic stage (after 3 weeks), the SDH contains iron oxidation products (ferritin and hemosiderin) which are paramagnetic, exhibiting high magnetic susceptibility. If rebleeding has occurred, the SDH demonstrates hypointense signal on T2* gradient recalled echo (GRE) and susceptibility-weighted imaging (SWI) sequences. In this chronic stage, T1WI shows hypointensity and T2WI shows hyperintensity of the cSDH. The difference in MR signal properties between intraparenchymal and extra-axial hemorrhage becomes most apparent in the chronic stage (>3 weeks) (see Table 18.2) (36, 47). It is of interest to the radiologist and perplexing to observe that a large cSDH without recent rebleeding may appear hyperintense on GRE and SWI sequences (e.g., lacking the expected susceptibility-induced-hypointensity) when other MRI sequences and/or antecedent serial imaging has shown imaging features to be consistent with a cSDH (Fig. 18.5).
 Figure 18.5 Chronic SDH and GRE MRI failing to demonstrate expected hemorrhage blooming. Serial CT and MRI in a six-month-old victim of prior AHT (shaking) who at initial presentation (at two months of age) showed bilateral mixed attenuation SDHs by NECT. As a result of a growing right supratentorial SDH, burr hole drainage was performed (within 24 hours of this MRI) and demonstrated petroleum-like qualities of a classic chronic SDH. A, Axial T1WI showing right holocranial SDH, hyperintense to CSF and cerebral cortex. B, Axial T2WI shows a hyperintense SDH and linear internal membrane (arrow) C, Axial GRE shows a hyperintense SDH, without hemorrhage blooming.
Figure 18.5 Chronic SDH and GRE MRI failing to demonstrate expected hemorrhage blooming. Serial CT and MRI in a six-month-old victim of prior AHT (shaking) who at initial presentation (at two months of age) showed bilateral mixed attenuation SDHs by NECT. As a result of a growing right supratentorial SDH, burr hole drainage was performed (within 24 hours of this MRI) and demonstrated petroleum-like qualities of a classic chronic SDH. A, Axial T1WI showing right holocranial SDH, hyperintense to CSF and cerebral cortex. B, Axial T2WI shows a hyperintense SDH and linear internal membrane (arrow) C, Axial GRE shows a hyperintense SDH, without hemorrhage blooming. The explanation of this phenomenon is based upon the physical principle that the absence of the blood brain barrier (BBB) within the extra-axial compartment promotes the resorption of paramagnetic hemosiderin and ferritin from the cSDH, thus in time mitigating the local field distortions or blooming (e.g., hypointensity). This is in striking contrast to a cerebral parenchymal hematoma, which will leave residual hemosiderin behind for an extended period of time. In the setting of recurrent subdural compartment hemorrhage (re-bleeding), there is blooming on GRE and SWI due to the presence of acute and subacute blood products.
In Fig. 18.5, note that the T1WI and T2WIs show signal features that suggest late subacute to early chronic right frontal hemorrhage. There is also an internal membrane within the right supratentorial SDH. The identification of membrane structure within a SDH is a marker of cSDH (see the cSDH section below for further discussion). Also note the smaller thin left frontal supratentorial SDH in Figure 18.5. There is bilateral expansion of the frontal SASs showing T2 signal intensity slightly greater than cerebral cortex.
Intracranial extra-axial hemorrhage: focused discussion
In this section, discussion will focus upon intracranial extra-axial hemorrhages including: epidural, subarachnoid, subdural, intraventricular, and subpial. Parenchymal hemorrhage and injury will be discussed in Chapter 19.
Epidural hemorrhage
Although EDH is uncommon in children, it is a potentially lethal complication of head trauma. It represents a collection of blood between the inner calvarium and outer periosteal/endosteal dura. Focal impact to the skull imparting a translational linear force (coup injury) is a typical cause of EDH (Fig. 18.6) (16).
 Figure 18.6 Acute arterial EDH. A, Coronal graphic shows a large biconvex acute EDH medially displacing the periosteal dura and brain. The swirling internal architecture of the hemorrhage indicates active bleeding. The overlying depressed calvarial fracture depicts the likely cause of laceration of the middle meningeal artery. Note the associated subfalcine herniation. B, Three-year-old who fell from a second-story window onto concrete. NECT coronal reformation shows large left acute EDH with a mixed attenuation, the “swirl sign.” Bleeding from the posterior branch of the middle meningeal artery was identified at time of hematoma evacuation. Graphic used with permission from Amirsys, Inc., Osborn’s Brain: Imaging. Pathology and Anatomy, 2012.
Figure 18.6 Acute arterial EDH. A, Coronal graphic shows a large biconvex acute EDH medially displacing the periosteal dura and brain. The swirling internal architecture of the hemorrhage indicates active bleeding. The overlying depressed calvarial fracture depicts the likely cause of laceration of the middle meningeal artery. Note the associated subfalcine herniation. B, Three-year-old who fell from a second-story window onto concrete. NECT coronal reformation shows large left acute EDH with a mixed attenuation, the “swirl sign.” Bleeding from the posterior branch of the middle meningeal artery was identified at time of hematoma evacuation. Graphic used with permission from Amirsys, Inc., Osborn’s Brain: Imaging. Pathology and Anatomy, 2012.Pediatric EDHs most often result from laceration of meningeal arterial vessels, dural venous sinus, or diploic veins. Venous EDH is more common among younger pediatric patients, whereas arterial EDHs are found more frequently in older children, adolescents, and adults (5, 7, 25). Unique anatomic and developmental features of the dura and calvarium of the infant and young child are important to consider. There is a greater pliancy of the pediatric calvarium compared to the adult. Therefore, it is not surprising that the prevalence of skull fracture associated with EDH in children is much less than a nearly 90% fracture prevalence reported among adults with EDH (56). Also, the skull of the young pediatric patient normally demonstrates shallow arterial grooves along the inner table of the calvarium in which the meningeal artery and its branches run. With head trauma, the artery is more mobile than in adults and less likely to be injured. Additionally, when compared to the adult, the pediatric periosteal dura is more firmly attached to the inner table of the skull. When arterial injury does occur, the posterior branch of the middle meningeal artery is the most common vessel injured (57–59).
Pediatric EDH is much less common than SDH and SAH following head injury (60). Duhaime and colleagues evaluated 100 children under 2 years of age suffering from head trauma (accidental and inflicted). Three children had EDHs, all from witnessed falls less than 120 cm in height (61). Merten et al. detected 2 EDHs among 47 pediatric AHT victims (62). Uncommon nontraumatic causes of EDH include hemorrhage following craniotomy or craniectomy, rare arteriovenous malformations (AVMs) of the dura or calvarium, invasive infections, neoplasms, and neoplastic-like disorders invading the dura and/or inner table of the skull (56). Rapid clinical assessment, accurate diagnosis, and appropriate management remain critical to avoid mortality and minimize morbidity in the setting of EDH.
The location of the EDH gives the radiologist a clue as to whether the hemorrhage is arterial or venous in origin. EDH in the temporal, frontoparietal, or temporoparietal locations is usually arterial and commonly arises from injury to a branch of the middle meningeal artery. Huisman and Tschirch found that among their 57 pediatric patients with EDH, 43% were parietal and 38% frontal (63). Posterior fossa and paratentorial EDHs are usually venous in origin arising from the transverse venous sinus, calvarial diploe, or dura (64). EDH from injury to the sphenoparietal venous sinus (sinus of Breschet) leads to venous EDH within the middle cranial fossa anterior to the temporal lobe. This characteristic middle cranial fossa location of venous EDH is often observed in the setting of facial trauma and is commonly seen with a sphenotemporal buttress fracture (65). Venous EDHs are more likely than arterial hemorrhages to be managed nonoperatively because of their slower accumulation under lower pressure (Fig. 18.7) (Table 18.3) (57).
 Figure 18.7 Venous EDH. Two-year-old child presenting with occipital swelling and irritability. No history of trauma. A, Axial NECT shows low attenuation retrotorcular epidural collection (arrow). B, Sagittal IV contrast-enhanced CT (CECT) demonstrates an epidural collection (arrow) that extends above and below the tentorium, displacing the torcular Herophili ventrally. Note the occipital rim enhancing subperiosteal collection. C, Surface rendered, 3D CT shows complex occipital fractures (arrows) with a diastatic component. At surgery, chronic hematoma was evacuated. Other findings supported a diagnosis of inflicted injury.
Figure 18.7 Venous EDH. Two-year-old child presenting with occipital swelling and irritability. No history of trauma. A, Axial NECT shows low attenuation retrotorcular epidural collection (arrow). B, Sagittal IV contrast-enhanced CT (CECT) demonstrates an epidural collection (arrow) that extends above and below the tentorium, displacing the torcular Herophili ventrally. Note the occipital rim enhancing subperiosteal collection. C, Surface rendered, 3D CT shows complex occipital fractures (arrows) with a diastatic component. At surgery, chronic hematoma was evacuated. Other findings supported a diagnosis of inflicted injury. | Arterial (classic) EDH |
|
| Venous EDH
Posterior fossa
Anterior middle fossa Vertex
Clival |
|
Typically, the cranial sutures limit the extent of the EDH. However, there are important exceptions to this rule. At the location of the sagittal suture, the periosteal dura forms the outer wall of the superior sagittal dural venous sinus, thus allowing an EDH to cross midline. EDH that crosses a suture may also result from laceration of the superior sagittal venous sinus. Huisman and Tschirch described 57 pediatric patients with traumatic EDH (see Figs. 18.6B, 18.8); 11% demonstrated EDHs that crossed cranial sutures. The predisposing factor(s) were traumatic sutural diastasis and/or fracture crossing a suture. As the hematoma crosses the plane of the suture, the hematoma may exhibit a “waist.” It is important to communicate to the treating neurosurgeon the size, extent, and “waist” shape of the suture crossing EDH to insure effective management (63).
The clinical presentation of traumatic EDH is variable depending upon size and location of the hemorrhage, whether it is arterial or venous, the presence of associated brain injuries, and the age of the patient. A lucid interval following an initial brief loss of consciousness is not uncommon. Nausea, vomiting, headache, cranial nerve palsies, somnolence, and coma are common (66). Roughly 10–25% of pediatric arterial EDHs will demonstrate progressive expansion with neurologic deterioration. This possibility for progressive expansion and clinical deterioration is justification for intensive monitoring and serial NECT imaging in the 24–36 hours following trauma. Fortunately, with rapid detection and prompt recognition and treatment, an excellent outcome can generally be anticipated (67–69).
Imaging considerations
Imaging of EDH shows a lens-shaped (lentiform) or biconvex hemorrhage of varied attenuation (NECT) or signal intensity (MRI). NECT is the imaging study of choice for rapid initial imaging and triage. Soft tissue and bone algorithms are necessary. The acute arterial EDH typically shows a lentiform shape with high attenuation components (clotted blood) and low attenuation components (active bleeding versus clot and serum separation) on NECT. This “swirl sign” on NECT is observed in approximately one-third of arterial EDHs and is an indication of active bleeding (70, 71). Depending on the size of the EDH, the adjacent SAS is compressed and a buckling of the adjacent cerebral or cerebellar cortex is seen. Associated herniation secondary to mass effect is not uncommon (Fig. 18.8) (72–75).
 Figure 18.8 Acute traumatic EDH. Three-year-old child who fell from a one-story roof top to a concrete driveway. A, B, Axial and coronal (reformation) NECT images demonstrate a large heterogeneous right EDH with associated subfalcine herniation. A subtle linear nondisplaced fracture of the parietal bone (not shown) involved the course of the posterior branch of the middle meningeal artery. Active meningeal arterial bleeding was detected at the time of surgery. EDH crosses the plane of the right squamosal suture.
Figure 18.8 Acute traumatic EDH. Three-year-old child who fell from a one-story roof top to a concrete driveway. A, B, Axial and coronal (reformation) NECT images demonstrate a large heterogeneous right EDH with associated subfalcine herniation. A subtle linear nondisplaced fracture of the parietal bone (not shown) involved the course of the posterior branch of the middle meningeal artery. Active meningeal arterial bleeding was detected at the time of surgery. EDH crosses the plane of the right squamosal suture. MRI evaluation of acute EDHs typically shows the hematoma to be isointense on T1WI and T2WI. In the subacute stage, the hemorrhage shows T1 hyperintensity and T2 hypointensity. The displaced dura may be detected medial to the clot and adjacent to the compressed brain and shows T1 and T2 hypointensity (57, 58, 64, 76).
In addition to fully characterizing the EDH, the radiologist is responsible to assess for comorbid findings such as contralateral SDH, traumatic SAH, and brain injury (contusion, hemorrhage, diffuse axonal injury [DAI], edema, ischemia, and herniation).
There are uncommon causes of nontraumatic hemorrhage or tumefactions in the pediatric epidural space mimicking hemorrhage including: neoplasms (e.g., leukemia/lymphoma, primary neoplasms of the calvarium, and metastases), neoplastic-like disease (Langerhans cell histiocytosis), infection (e.g., fungal disease, dural tuberculosis, and other invasive bacteria), and extramedullary hematopoiesis. All of these conditions could present with intrinsic increased attenuation material within the epidural space on NECT. However, many of these rare considerations demonstrate bone destruction and/or contrast enhancement of the “epidural mass” (6).
Subarachnoid hemorrhage
Subarachnoid hemorrhage (SAH) is bleeding into the SAS bordered inwardly by the pia and outwardly by the arachnoid mater. Traumatic SAH represents a marker of brain displacement by angular brain movement, a result of acceleration–deceleration forces with or without associated impact injury (Fig. 18.9) (18, 77–79). In the pediatric population, trauma is the most common cause of SAH. Furthermore, SAH is more commonly associated with AHT than with AccT (Table 18.4) (80, 81).
 Figure 18.9 Traumatic SAH. A, Coronal graphic shows SAH within the left lateral cerebral or Sylvian fissure, adjacent to contused gyri, and within the suprasellar cistern. Note hemorrhage filling the frontal horns of the lateral ventricles and the third ventricle. Numerous parenchymal hemorrhagic contusions are also depicted. B, Coronal GRE MR image in a 10-week-old child with seizures, and macrocrania, shows hypointense (blooming) SAH (arrow). Note the bilateral peripheral holocranial hyperintense fluid. Other MR sequences were consistent with chronic SDHs. The other findings that supported inflicted injury were facial bruising and numerous CMLs. (A, With permission from Amirsys Publishing, Inc. Figure from Osborn AG. Osborn’s Brain: Imaging, Pathology and Anatomy. Baltimore, MD: Lippincott Williams & Wilkins; 2012.)
Figure 18.9 Traumatic SAH. A, Coronal graphic shows SAH within the left lateral cerebral or Sylvian fissure, adjacent to contused gyri, and within the suprasellar cistern. Note hemorrhage filling the frontal horns of the lateral ventricles and the third ventricle. Numerous parenchymal hemorrhagic contusions are also depicted. B, Coronal GRE MR image in a 10-week-old child with seizures, and macrocrania, shows hypointense (blooming) SAH (arrow). Note the bilateral peripheral holocranial hyperintense fluid. Other MR sequences were consistent with chronic SDHs. The other findings that supported inflicted injury were facial bruising and numerous CMLs. (A, With permission from Amirsys Publishing, Inc. Figure from Osborn AG. Osborn’s Brain: Imaging, Pathology and Anatomy. Baltimore, MD: Lippincott Williams & Wilkins; 2012.)| Features | AHT | AccT |
|---|---|---|
| Clinical | ||
| • Relevant history | –/+ | +++ |
| • Age of patient | Younger | Older |
| • Signs of impact | –/+ | +++ |
| • Severe retinal hemorrhage | +++ | – |
| Imaging findings | ||
| Extra-axial hemorrhage | ||
| • SDH | +++ | + |
| • SAH | +++ | + |
| • EDH | –/+ | +++ |
| Parenchymal injury | ||
| • Ischemia/infarction | +++ | –/+ |
| • Laceration | ++ | – |
| • Axonal injury (DAI) | –/+ | +++ |
Scalp swelling is significantly less common in AHT victims with the exception of fatal AHT cases.
In their review of NECT in a pediatric population with AHT, Cohen and colleagues observed SAH in 73% (80). Reece and Sege observed SAH in 70% of pediatric traumatic brain injury (TBI) patients who also had SDH. Subarachnoid hemorrhage in their series was more common among AHT victims (31%) than those individuals suffering AccT (8%) (82, 83). Duhaime et al. reported a high incidence of SAH in pediatric patients who suffered AHT in comparison to victims of AccT where SAH was only detected among those who experienced moderate to severe head trauma (e.g., high-speed motor vehicle accidents [MVAs]) (61). Duhaime, Jenny, and others have independently showed the common association between SAH and SDH in AHT victims (3, 21, 22, 84–90).
The tearing of cortical, pial, and arachnoid veins and less commonly arteries/arterioles from acceleration, deceleration forces represents a well-recognized and common source of SAH. Arachnoid vessels closely relate to the cortical dural bridging veins and, therefore, when arachnoid vessels are torn, bridging veins are also commonly torn, leading to the combination of SAH and SDH (Fig. 18.10).
 Figure 18.10 Traumatic SAH and SDH in the setting of AHT. Twenty-one-month-old child who returned from the babysitter lethargic and irritable. A, Axial NECT shows focal high attenuation (e.g., acute/early subacute) right frontal SAH (arrow). Note the high attenuation posterior paratorcular SDH (arrowhead). Peripheral frontotemporal chronic SDHs show attenuation greater than CSF. B, Axial T1WI demonstrates the hypointense acute paratorcular SDH (arrowhead) and shows the frontotemporal chronic SDHs to have signal intensity greater than ventricular CSF (arrow). C, Axial T2WI shows hyperintense bilateral frontotemporal chronic SDHs and heterogeneous T2 hypointensity of the paratorcular aSDH. D, Axial T2 FLAIR sequence image shows the sharp discrimination between the peripheral hypointense frontotemporal chronic SDHs and the adjacent mildly expanded SASs. Note the similarity of signal intensity between the subarachnoid and ventricular CSF. E, F, Axial SWI and coronal GRE images demonstrate hypointensity of acute/early subacute SAH (arrow). Note the hyperintensity of the peripheral chronic SDHs. There were other findings supporting abuse.
Figure 18.10 Traumatic SAH and SDH in the setting of AHT. Twenty-one-month-old child who returned from the babysitter lethargic and irritable. A, Axial NECT shows focal high attenuation (e.g., acute/early subacute) right frontal SAH (arrow). Note the high attenuation posterior paratorcular SDH (arrowhead). Peripheral frontotemporal chronic SDHs show attenuation greater than CSF. B, Axial T1WI demonstrates the hypointense acute paratorcular SDH (arrowhead) and shows the frontotemporal chronic SDHs to have signal intensity greater than ventricular CSF (arrow). C, Axial T2WI shows hyperintense bilateral frontotemporal chronic SDHs and heterogeneous T2 hypointensity of the paratorcular aSDH. D, Axial T2 FLAIR sequence image shows the sharp discrimination between the peripheral hypointense frontotemporal chronic SDHs and the adjacent mildly expanded SASs. Note the similarity of signal intensity between the subarachnoid and ventricular CSF. E, F, Axial SWI and coronal GRE images demonstrate hypointensity of acute/early subacute SAH (arrow). Note the hyperintensity of the peripheral chronic SDHs. There were other findings supporting abuse. Uncommon causes of pediatric SAH include: peripheral brain parenchymal hematoma that ruptures into the SAS, ventricular hemorrhage originating from the choroid plexus or ventricular ependyma, and nontraumatic arterial sources (e.g., cerebral arterial aneurysm and cerebral/spinal vascular malformations). Arterial dissection is another important cause of SAH that may follow head, craniocervical junction, and neck trauma (3).
The symptoms and signs accompanying SAH are often a reflection of associated brain injury (e.g., contusion, axonal injury, ischemia, and infarction). Blood in the SAS is a cortical irritant, and a known cause of seizures. Delayed complications of SAH include vasospasm with resultant ischemia and infarction, and post-hemorrhagic hydrocephalus. A rare delayed complication of trauma is the formation of a post-traumatic pseudoaneurysm with resultant delayed SAH (5, 16, 91, 92). Cerebral CT angiography (CTA) represents an important imaging study in evaluating SAH, particularly when hemorrhage volume and location are out of proportion to the history of trauma, or when SAH prevails in the absence of SDH.
Imaging considerations
With the exception of location, the imaging appearance of traumatic SAH is similar to aneurysmal SAH or hemorrhage resulting from vessel dissection and rupture. SAH hugs the surface of the brain, interdigitating into sulci and filling cisterns. The common intracranial locations of traumatic SAH include the lateral cerebral or sylvian fissures; anteroinferior frontal and temporal SASs, cerebral hemispheric convexity sulci, interhemispheric fissure, interpeduncular fossa, suprasellar cistern, and basal cisterns (Figs. 18.11, 18.12) (93).
 Figure 18.11 Traumatic SAH. A, Axial graphic through the upper brainstem shows SAH filling the suprasellar cistern, lateral cerebral and interpeduncular cisterns. B, Axial NECT in a 2-year-old victim of a pedestrian MVA shows copious high attenuation SAH within the basal cisterns and fourth ventricle. Note the early post-hemorrhagic hydrocephalus as evidenced by temporal horn and fourth ventricular dilation. Color graphic used with permission from Amirsys Publishing, Inc. Figure from Osborn AG. Osborn’s Brain: Imaging, Pathology and Anatomy. Baltimore, MD: Lippincott Williams & Wilkins; 2012.
Figure 18.11 Traumatic SAH. A, Axial graphic through the upper brainstem shows SAH filling the suprasellar cistern, lateral cerebral and interpeduncular cisterns. B, Axial NECT in a 2-year-old victim of a pedestrian MVA shows copious high attenuation SAH within the basal cisterns and fourth ventricle. Note the early post-hemorrhagic hydrocephalus as evidenced by temporal horn and fourth ventricular dilation. Color graphic used with permission from Amirsys Publishing, Inc. Figure from Osborn AG. Osborn’s Brain: Imaging, Pathology and Anatomy. Baltimore, MD: Lippincott Williams & Wilkins; 2012. Figure 18.12 Traumatic SAH. A, Axial color graphic illustrates left convexal SAH (purple). B, Axial T2 FLAIR image in a 6-year-old child who fell from her bike without a helmet, shows acute hyperintense SAH within bilateral parietal sulci (arrows). (A, With permission from Amirsys Publishing, Inc. Figure from Osborn AG. Osborn’s Brain: Imaging, Pathology and Anatomy. Baltimore, MD: Lippincott Williams & Wilkins; 2012)
Figure 18.12 Traumatic SAH. A, Axial color graphic illustrates left convexal SAH (purple). B, Axial T2 FLAIR image in a 6-year-old child who fell from her bike without a helmet, shows acute hyperintense SAH within bilateral parietal sulci (arrows). (A, With permission from Amirsys Publishing, Inc. Figure from Osborn AG. Osborn’s Brain: Imaging, Pathology and Anatomy. Baltimore, MD: Lippincott Williams & Wilkins; 2012)The presence of SAH within the interpeduncular fossa following trauma may be a marker for associated brainstem injury (Fig. 18.13) (16). On NECT, distinguishing SAH from focal subpial hemorrhage or hemorrhagic cortical contusion may be difficult. The MRI fluid-attenuated inversion recovery (FLAIR) sequence is particularly helpful to detect SAH (Fig. 18.12B) (94). Traumatic SAH should always be considered a proxy for underlying brain injury (95).
 Figure 18.13 Traumatic SAH; a proxy for axonal injury. A, Sagittal graphic depicts the common locations of axonal injury including the corpus callosum and brainstem. Note the traumatic intraventricular and diffuse SAH layering dependently. B, Sagittal GRE image shows hypointense hemorrhage within the cerebral aqueduct, partially obstructing the third ventricle (arrow). Graphic used with permission from Amirsys Publishing, Inc. Figure from Osborn AG. Osborn’s Brain: Imaging, Pathology and Anatomy. Baltimore, MD: Lippincott Williams & Wilkins; 2012.
Figure 18.13 Traumatic SAH; a proxy for axonal injury. A, Sagittal graphic depicts the common locations of axonal injury including the corpus callosum and brainstem. Note the traumatic intraventricular and diffuse SAH layering dependently. B, Sagittal GRE image shows hypointense hemorrhage within the cerebral aqueduct, partially obstructing the third ventricle (arrow). Graphic used with permission from Amirsys Publishing, Inc. Figure from Osborn AG. Osborn’s Brain: Imaging, Pathology and Anatomy. Baltimore, MD: Lippincott Williams & Wilkins; 2012.Nontraumatic causes of SAH
Nontraumatic causes of SAH are rare in infants and young children. The location of SAH within the anterior interhemispheric fissure is observed with anterior communicating artery aneurysm rupture, and subarachnoid blood within the suprasellar cistern may be seen following rupture of a posterior communicating artery aneurysm. This suprasellar location of hemorrhage may also be seen following traumatic rupture of the posterior communicating artery. SAH is often detected in the basal cisterns, foramen of Magendie, and foramina of Lushka. A predominantly basal distribution of SAH may originate from a posterior inferior cerebellar artery aneurysm, spinal AVM, or a hemorrhagic spinal cord tumor (e.g., conus or filal myxopapillary ependymoma) (Fig. 18.14) (6, 96).
 Figure 18.14 Aneurysmal SAH. A, B, Axial NECT images in a six-week-old infant with new-onset generalized seizures and respiratory arrest at home which required initiation of CPR shows copious SAH (arrows). Note SAH within the lateral cerebral fissures (arrows) and fourth ventricle. No SDH was identified. C, Sagittal T2WI depicts a large intradural peri-conal aneurysmal flow void (arrow). Spinal angiography confirmed a large venous varix. The referring hospital’s radiologist had interpreted the diffuse SAH to be a result of AHT. The patient was subsequently diagnosed with hereditary hemorrhagic telangiectasia.
Figure 18.14 Aneurysmal SAH. A, B, Axial NECT images in a six-week-old infant with new-onset generalized seizures and respiratory arrest at home which required initiation of CPR shows copious SAH (arrows). Note SAH within the lateral cerebral fissures (arrows) and fourth ventricle. No SDH was identified. C, Sagittal T2WI depicts a large intradural peri-conal aneurysmal flow void (arrow). Spinal angiography confirmed a large venous varix. The referring hospital’s radiologist had interpreted the diffuse SAH to be a result of AHT. The patient was subsequently diagnosed with hereditary hemorrhagic telangiectasia. In the setting of acute trauma or nontraumatic acute neurologic deterioration, NECT is the imaging study of choice. CT shows acute/subacute SAH as linear high attenuation fluid (relative to brain parenchyma) within the sulci. SAH within the intracranial cisterns conforms to the shape of the cisternal space(s). Victims of AHT commonly show accompanying SDH. Detecting SAH on routine MR sequences can be challenging as acute hemorrhage within the SAS is nearly isointense to gray matter on T1WI and T2WI. A smudged or “dirty” interface at sulcal–gyral interfaces is a clue to the presence of SAS hemorrhage. SAH shows sulcal hyperintensity on T2 FLAIR MRI (as shown in Fig. 18.12B). The utilization of GRE and SWI sequences, both of which exploit the magnetic susceptibility properties of oxidizing hemorrhage, heightens the sensitivity of detecting SAH. The age of hemorrhage and occurrence of re-bleeding affect the presence and quantity of hemosiderin and ferritin within the SAH and SAS and will dictate whether local MR field distortions (hypointensities) prevail on GRE and SWI sequences (48, 49, 70, 97–101). The presence of acute SAH on cranial sonography manifests as hyperechoic widening of the cortical sulci (102).
Pseudo-SAH
Pseudo-SAH is an observation made on NECT and mimics SAH. It results when the pathologic low attenuation of the brain (e.g., diffuse cerebral hemispheric edema) contrasts with the relative increased attenuation of the congested pial venous structures and the hyperattenuating appearance of cisternal arteries and veins. This is particularly prominent within the suprasellar cistern. The key to suspecting this diagnosis on NECT is noting that the suprasellar region of increased attenuation is smooth, conforms to vessels and that there is a conspicuous lack of hemorrhage in typical SASs (e.g., interpeduncular cistern, interhemispheric and sylvian fissures) (Fig. 18.15). MRI with GRE and SWI show an absence of hemorrhage dephasing (hypointensity) in cases of suspected pseudo-SAH (5, 16).
 Figure 18.15 Pseudo-SAH. Axial NECT in a two-month-old infant following cardiopulmonary arrest and subsequent status epilepticus demonstrates high attenuation within the suprasellar cistern (arrow). Note the obliteration of the basal cisterns secondary to surrounding diffuse cerebral edema. There was no hemorrhage identified within the lateral cerebral fissures or ventricles. Autopsy revealed no SAH.
Figure 18.15 Pseudo-SAH. Axial NECT in a two-month-old infant following cardiopulmonary arrest and subsequent status epilepticus demonstrates high attenuation within the suprasellar cistern (arrow). Note the obliteration of the basal cisterns secondary to surrounding diffuse cerebral edema. There was no hemorrhage identified within the lateral cerebral fissures or ventricles. Autopsy revealed no SAH. In addition to pseudo-SAH, other potential mimics of SAH include T2 FLAIR sulcal hyperintensity observed in the following settings: secondary to MRI artifact from incomplete CSF suppression, associated with increased inspired oxygen during general anesthesia, effect of intravenous (IV) propofol sedation, and contrast leakage into the SAS with renal failure and SAS metastatic disease (16). Very cellular tumors with high nuclear-to-cytoplasmic ratios (e.g., leukemia, primitive neuroectodermal tumor [PNET], germinoma, and melanoma) as well as pyogenic infections may cause sulcal hyperattenuation on NECT (6).
Subdural hemorrhage
SDH represents the most common observed intracranial abnormality in infants and young children who have suffered AHT (7, 20, 26, 28). A spectrum of clinical symptoms and signs may be found in children with SDH. This includes irritability, lethargy, altered mental status, loss of consciousness, seizures, vomiting, and signs of increased intracranial pressure (e.g., bulging fontanel, and papilledema). Approximately half of the youngest children with SDH present with a significantly depressed Glasgow Coma Score (GCS) or coma (35, 103). Mortality in pediatric patients presenting with SDH ranges from 10 to 20% (22, 34, 78, 81, 104–106). In the 1970s the contributions of Guthkelch and Caffey describing infants who suffered whiplash shaking raised awareness that caretaker shaking could inflict serious and even fatal head injury, including the presence of SDH (107–110). The biomechanical underpinnings to these early clinical observations are that acceleration–deceleration forces without or with head impaction cause the intracranial contents (e.g., gray matter, white matter, vessels, dural structures, and bone) to move at different velocities (see Chapter 16) (111–116).
SDH is much more common than EDH and represents an important cause of death and disability following significant head trauma. Acute SDH in the infant is a cardinal feature of AHT, representing the most common intracranial imaging abnormality detected among those suffering inflicted trauma (78, 117–122). The detection of SDH of different ages without a substantiating health history is strongly suggestive of AHT (Fig. 18.16) (7, 21, 22, 78, 81, 90, 106).
 Figure 18.16 SDHs of different ages. A, Coronal graphic depicts an acute right holocranial SDH (purple) with associated mass effect and right to left subfalcine herniation. There is a smaller left SDH of an older age (yellow). Note the associated SAH (arrows) and peripheral hemorrhagic contusions (arrowheads). B, An eight-month-old infant with patterned bruising and skeletal injuries supporting inflicted trauma. Coronal T2WI demonstrates a loculated or membrane bound early subacute right frontal (arrow) and large chronic left holocranial SDH (arrowhead). (A, With permission from Amirsys Publishing, Inc. from Osborn AG. Osborn’s Brain: Imaging, Pathology and Anatomy. Baltimore, MD: Lippincott Williams & Wilkins; 2012.)
Figure 18.16 SDHs of different ages. A, Coronal graphic depicts an acute right holocranial SDH (purple) with associated mass effect and right to left subfalcine herniation. There is a smaller left SDH of an older age (yellow). Note the associated SAH (arrows) and peripheral hemorrhagic contusions (arrowheads). B, An eight-month-old infant with patterned bruising and skeletal injuries supporting inflicted trauma. Coronal T2WI demonstrates a loculated or membrane bound early subacute right frontal (arrow) and large chronic left holocranial SDH (arrowhead). (A, With permission from Amirsys Publishing, Inc. from Osborn AG. Osborn’s Brain: Imaging, Pathology and Anatomy. Baltimore, MD: Lippincott Williams & Wilkins; 2012.)When considered in isolation, the observation of SDH lacks some specificity in distinguishing inflicted from accidental trauma. However, in a pre-mobile infant, in the absence of a credible history or when SDH is identified in association with other findings including: (1) bruising; (2) severe retinal hemorrhages (RHs); (3) multiple sites and ages of intracranial hemorrhage; (4) cerebral parenchymal abnormalities (e.g., edema, ischemia, infarction, or parenchymal laceration); (5) injury to the cranial cervical junction; and if there are no features to suggest a nontraumatic cause of SDH, then SDH is strongly predictive of AHT (117, 120, 123–125).
Over a five-year period, Pollanen and colleagues chronicled child abuse deaths in Ontario, Canada. Of the 21 deaths confirmed as child abuse–maltreatment syndrome, 50% were less than 6 months of age and 71% had evidence of AHT, with SDH being the most prevalent finding. Additionally, in their report, most cases of fatal shaking injury demonstrated evidence of previous head injury (124). Morris and colleagues concluded that the detection of SDH in children in the absence of a specific incident such as an auto accident or a medical condition, for example, a bleeding diathesis, “raises significant concern that child abuse is the diagnosis” (126). Finally, Billmire and Myers state: “ICI in children less than one year of age without an explanation constitutes grounds for child abuse investigation” (127).
Neuropathologic and neuroimaging considerations
A subdural compartment hemorrhage or hematoma is blood that lies interposed between the inner dura and the arachnoid mater (7, 16, 32, 128–131). SDH is crescent or concavoconvex in shape. The hemorrhage effaces adjacent gyri and sulci, does not extend into sulci, displaces cortical dural veins inwardly against the surface of the brain, and, if large enough, compresses the adjacent brain causing herniation (Fig. 18.17).
 Figure 18.17 Acute SDH with brain compression. A, Axial graphic shows an acute holocranial SDH compressing the left cerebral hemisphere, causing left-to-right subfalcine herniation. Note the numerous scattered hemorrhagic parenchymal contusions. B, Axial NECT in a 19-month-old victim of confessed shaking shows a left hemispheric mixed attenuation SDH (arrow) with early subfalcine herniation. Note the anterior and posterior high attenuation components of the falcine SDH (arrowheads). Surgery confirmed an aSDH and an edematous left cerebral hemisphere. Multiple rib and metaphyseal fractures were detected on the SS. (A, With permission from Amirsys Publishing, Inc. from Osborn AG. Osborn’s Brain: Imaging, Pathology and Anatomy. Baltimore, MD: Lippincott Williams & Wilkins; 2012.)
Figure 18.17 Acute SDH with brain compression. A, Axial graphic shows an acute holocranial SDH compressing the left cerebral hemisphere, causing left-to-right subfalcine herniation. Note the numerous scattered hemorrhagic parenchymal contusions. B, Axial NECT in a 19-month-old victim of confessed shaking shows a left hemispheric mixed attenuation SDH (arrow) with early subfalcine herniation. Note the anterior and posterior high attenuation components of the falcine SDH (arrowheads). Surgery confirmed an aSDH and an edematous left cerebral hemisphere. Multiple rib and metaphyseal fractures were detected on the SS. (A, With permission from Amirsys Publishing, Inc. from Osborn AG. Osborn’s Brain: Imaging, Pathology and Anatomy. Baltimore, MD: Lippincott Williams & Wilkins; 2012.)Common locations of SDH include: the cerebral convexities, frontotemporal region, adjacent to or within the falx cerebri (particularly the anteroinferior and posterior falx), tentorium cerebelli, and middle and posterior cranial fossae. SDH may cross the plane of cranial sutures but remains limited by several normal anatomic structures including the falx cerebri, tentorium cerebelli, and dural venous sinuses (9, 15, 28, 79, 132–135).
Recognizing the role of the intracranial dural barriers (e.g., falx cerebri and tentorium cerebelli) to limit SDH migration has important forensic implications, as there are experts who posit and testify to the fact that SDH may naturally migrate between supratentorial and infratentorial compartments (3, 9, 136). There is no evidence to support such an assertion. Routine sagittal and coronal NECT reconstructions and the multiplanar imaging capabilities of MRI aid routinely in characterizing and accurately compartmentalizing SDH (Fig. 18.18). In the author’s institution, sagittal and coronal reformations are added routinely to all NECT examinations in patients less than two years of age whatever the clinical indication for the study and in all head trauma patients no matter what age (9, 19, 20, 27, 28, 137, 138).
 Figure 18.18 SDH, value of NECT multiplanar reconstructions. Nonenhanced CT in an 11-week-old infant presenting with seizures and obtundation. A–C, Axial, coronal, and sagittal NECT images show a large, compressive, mixed attenuation left convexity and parafalcine SDH. Note the subfalcine herniation and loss of gray–white differentiation indicative of diffuse left cerebral hemispheric edema. There is a small right parafalcine SDH best seen on the coronal image. At surgery, a tense left hyperacute and aSDH was evacuated and two bleeding torn bridging veins were coagulated. Torso and facial bruising was also observed. The perpetrator confessed to shaking the infant.
Figure 18.18 SDH, value of NECT multiplanar reconstructions. Nonenhanced CT in an 11-week-old infant presenting with seizures and obtundation. A–C, Axial, coronal, and sagittal NECT images show a large, compressive, mixed attenuation left convexity and parafalcine SDH. Note the subfalcine herniation and loss of gray–white differentiation indicative of diffuse left cerebral hemispheric edema. There is a small right parafalcine SDH best seen on the coronal image. At surgery, a tense left hyperacute and aSDH was evacuated and two bleeding torn bridging veins were coagulated. Torso and facial bruising was also observed. The perpetrator confessed to shaking the infant. A rich literature exists to support acceleration–deceleration and angular momentum, without or with impact, as mechanisms predisposing to the development of traumatic SDH (see Chapter 16). With inflicted injury, the brain moves more than fixed structures (falx cerebri, tentorium cerebelli, dural venous sinuses, and rough bony prominences [pterion, floor of the anterior and middle cranial fossa]). The shearing forces created may cause a spectrum of injury including SDH, SAH, and brain parenchymal injuries, including cortical contusion, DAI, and occasionally parenchymal laceration (32, 104, 111, 113, 118, 129, 139, 140).
There is some debate as to whether a true subdural space exists and when SDH occurs, whether it accumulates within this space or is contained within the dura (e.g., intradural hemorrhage) (15). For the neuropathologist, there is no question that there is a potential subdural space distinct from the SAS, and that pathologic processes arising in each space (e.g., blood, pus, and tumor cells) tend to remain compartmentalized. At autopsy, when the skull cap is opened and lifted away, and the dura is cut, there is no apparent tear of the leptomeninges or flow of CSF from the subarachnoid space. The neuropathologist looking at the brain appreciates both the dural and arachnoid surfaces to be pristine and intact. At autopsy, SDH may be observed to have migrated extensively throughout the subdural space, moving in accordance with gravity, pooling in locations such as the middle cranial fossa, and posterior to the occipital lobes (limited by the falx cerebri and tentorium cerebelli). This diffuse unimpeded migration of SDH is more common in children than adults. The gravitational settling of hemorrhage yields a hematocrit or sediment effect on NECT and MRI, often arising from a single traumatic incident, and should not be misinterpreted as hemorrhages of differing ages. Upon evacuation of the SDH, the arachnoid mater is intact (pers. comm. with Lucy B. Rorke-Adams MD; 2, 3, 85, 105, 141–144).
To the electron microscopist and histopathologist, the potential subdural space is viewed differently. Orlin and colleagues described the electron microscopic features of the inner DBCL as a delicate vascular tissue with flake-like relatively electron lucent cells stacked upon each other with narrow intercellular clefts (14). Haines et al., in their electron microscopic observations, reported that the subdural space does not exist and, when created, it is done so either artifactually or as a result of pathologic conditions. Furthermore, Haines stated that this cleavage occurs at the DBCL (13, 145). Orlin et al., like Haines, reported that under normal conditions there is no normal subdural space and coined the term subdural compartment to describe the location where, following cleavage and disruption of the DBCL, hemorrhage, and nonhemorrhagic subdural fluid accumulates (14). Interestingly, Frederickson, in his histologic evaluation of the inner DBCL in guinea pigs determined that the DBCL varied in thickness based on location (thicker in the parafalcine region) and in some supratentorial areas (frontal lobes) was absent all together. Microscopic observations reveal that when the dura is separated from the arachnoid, cells from the DBCL were found on the arachnoid and the dural sides of the cleavage plane (146). More recently, Mack et al. have recapitulated the insights previously made by Haines et al., emphasizing their nomenclature, anatomic descriptions, and the histologic characterizations of the DBCL (4, 13–15, 145). Mack and colleagues highlight the intradural vascular plexus as a potential source of subdural compartment hemorrhage (Fig. 18.19) (15).
 Figure 18.19 Dural venous plexus. Coronal graphic shows the artist’s depiction of the dural venous plexus (DP) located within the inner portion of the meningeal dura. (BV bridging vein, MA meningeal artery, MV meningeal vein, PA penetrating arteriole, SS superior sagittal sinus). (From Mack J, Squier W, Eastman JT. Anatomy and development of the meninges: implications for subdural collections and CSF circulation. Pediatr Radiol. 2009;39:200–10, with kind permission from Springer Science and Business Media.)
Figure 18.19 Dural venous plexus. Coronal graphic shows the artist’s depiction of the dural venous plexus (DP) located within the inner portion of the meningeal dura. (BV bridging vein, MA meningeal artery, MV meningeal vein, PA penetrating arteriole, SS superior sagittal sinus). (From Mack J, Squier W, Eastman JT. Anatomy and development of the meninges: implications for subdural collections and CSF circulation. Pediatr Radiol. 2009;39:200–10, with kind permission from Springer Science and Business Media.)There is ample neurosurgical, neuroimaging, and neuropathologic evidence that traumatic SDH originates from torn bridging veins. These bridging veins extend from the cortical surface and drain into the dural venous sinuses, particularly the superior sagittal venous sinus (see Fig. 18.1). Bridging veins may tear as the brain moves under the acceleration–deceleration and rotational movements following trauma (147–150).
The dural entrance of the bridging veins is a focus of interest for many researchers who posit a continuum of shear strain, vein tearing, bleeding, and thrombosis occurring in the setting of trauma, particularly AHT (150–152). Numerous small cortical veins drain from the surface of the brain, traverse the SAS, and coalesce into larger bridging veins that ultimately drain into the superior sagittal venous sinus (see Figs. 18.1, 18.20). These bridging veins constitute a pipeline between the arachnoid and the dura. For the radiologist, localizing the position of the cortical veins represents an important landmark in distinguishing prominent SASs from subdural fluid/blood collections (see discussion in Benign enlargement of the SASs in infants section) (153–155).
 Figure 18.20 CTV demonstrating normal bridging veins. A–C, Axial CECT with coronal and sagittal reformations from intravenously enhanced CTV demonstrating normal contrast-enhanced bridging veins (arrowheads). Note how the bridging veins (white arrowheads) enter the superior sagittal venous sinus at an angle opposite to the direction of normal blood flow.
Figure 18.20 CTV demonstrating normal bridging veins. A–C, Axial CECT with coronal and sagittal reformations from intravenously enhanced CTV demonstrating normal contrast-enhanced bridging veins (arrowheads). Note how the bridging veins (white arrowheads) enter the superior sagittal venous sinus at an angle opposite to the direction of normal blood flow. The mean number of bridging veins per cerebral hemisphere is less than 15, ranging between 6 and 12 per hemisphere. The diameter of the bridging veins is inversely proportional to the number (152). In the adult, depending on the total number of bridging veins, the diameter may be 3–4 mm. In infants, the bridging veins are smaller (156–159). A cadaveric digital subtraction cerebral angiography study by Han and colleagues demonstrated that normal cortical bridging veins enter the superior sagittal venous sinus at an angle opposite to the direction of normal blood flow. This angle of insertion explains why bridging veins are more susceptible to disruption with occipital than frontal impact (Fig. 18.20) (151).
Yamashima and Friede showed with electron microscopy the thin wall of the subdural cuff of the bridging vein and the absent mural reinforcement as the vein penetrates the inner dura before entering the superior sagittal venous sinus, thus rendering the vein susceptible to disruption at this location as a result of trauma (160). The sagittal orientation of the falx cerebri confers some protection to the bridging veins with side-to-side stress and strain. In the setting of acceleration–deceleration forces, particularly in the anterior–posterior plane without or with impact, there is risk of bridging vein disruption. With tearing of the bridging vein, hemorrhage into the subdural and subarachnoid spaces is observed.
Adamsbaum and Rambaud have highlighted the important observation of torn and thrombosed bridging veins in a case series including one neonate and three infants with supporting clinical and forensic data, cross-sectional brain imaging (e.g., NECT and MRI), and autopsy correlation. All four patients died as a result of AHT (confirmed by confession) (Fig. 18.21). In their series, the authors draw attention to the cerebral convexities where on CT and MRI, traumatically disrupted and subsequently thrombosed bridging veins were identified as markers of the traumatic origin of SAH and SDHs (150). The bridging vein, traumatically torn from its dural mooring, will initially bleed and within hours undergo thrombosis. In the acute and early subacute stages, tubular- and lollipop-shaped high attenuation hemorrhages will be evident over the vertex on NECT. On MRI, these disrupted and thrombosed bridging veins appear hypointense on T2-weighted and GRE imaging and either isointense to hyperintense on T1WI depending upon the age of the hemorrhage (Figs. 18.22, 18.23).
 Figure 18.21 Bridging vein thrombosis. A, B, Autopsy specimens demonstrating tubular, torn, and thrombosed bridging veins (white arrowheads). Views are looking down upon the cerebral convexities at autopsy. Assailant admitted injury by shaking. (Courtesy of Dr. Caroline Rambaud.)
Figure 18.21 Bridging vein thrombosis. A, B, Autopsy specimens demonstrating tubular, torn, and thrombosed bridging veins (white arrowheads). Views are looking down upon the cerebral convexities at autopsy. Assailant admitted injury by shaking. (Courtesy of Dr. Caroline Rambaud.) Figure 18.22 Bridging vein thrombosis. Images in a five-month-old infant presenting with seizures, loss of consciousness, and multiple classic metaphyseal fractures. Probable multiple bridging vein thromboses (arrowheads), seen on GRE (T2*)-weighted MRI (A) and on axial CT (B). Chronic bi-frontal SDHs (hypodense) associated with hyperdense, acute interhemispheric SDH (C). (From Adamsbaum C, Rambaud C. Abusive head trauma: don’t overlook bridging vein thrombosis. Pediatr Radiol. 2012;42(11):1298–300, with kind permission from Springer Science and Business Media.)
Figure 18.22 Bridging vein thrombosis. Images in a five-month-old infant presenting with seizures, loss of consciousness, and multiple classic metaphyseal fractures. Probable multiple bridging vein thromboses (arrowheads), seen on GRE (T2*)-weighted MRI (A) and on axial CT (B). Chronic bi-frontal SDHs (hypodense) associated with hyperdense, acute interhemispheric SDH (C). (From Adamsbaum C, Rambaud C. Abusive head trauma: don’t overlook bridging vein thrombosis. Pediatr Radiol. 2012;42(11):1298–300, with kind permission from Springer Science and Business Media.) Figure 18.23 Probable convexal torn and thrombosed bridging veins. A, Axial NECT image in a 10-week-old infant who presented with seizures demonstrates convexity tubular high attenuation hemorrhage suspicious for thrombosed/torn bridging veins (arrows). Also note the high attenuation posterior parafalcine SDH (arrowheads). B, At 12 weeks of age, the GRE MRI through the convexities, demonstrates numerous tubular and rounded hypointense (“blooming”) hemorrhages compatible with torn and thrombosed bridging veins. The clinical circumstances were suspicious for AHT.
Figure 18.23 Probable convexal torn and thrombosed bridging veins. A, Axial NECT image in a 10-week-old infant who presented with seizures demonstrates convexity tubular high attenuation hemorrhage suspicious for thrombosed/torn bridging veins (arrows). Also note the high attenuation posterior parafalcine SDH (arrowheads). B, At 12 weeks of age, the GRE MRI through the convexities, demonstrates numerous tubular and rounded hypointense (“blooming”) hemorrhages compatible with torn and thrombosed bridging veins. The clinical circumstances were suspicious for AHT. Some authors posit that tearing of bridging veins is an unlikely cause of infant SDH as the bleeding would be massive owing to the large volumes of blood that are carried in the adult bridging veins to the superior sagittal venous sinus (112, 157, 158, 161). This opinion does not account for the smaller size of the infant’s bridging veins compared to the adult, contributions of the infant’s normal coagulation cascade to achieve thrombosis, and the tamponading effect of accumulating extra-axial hemorrhage and adjacent edematous brain, all collectively promoting thrombosis of the torn bridging vein.
In addition to cortical and bridging vein disruption resulting in subdural compartment hemorrhage, bleeding may originate within the dura at the inner DBCL (Fig. 18.24) (15).
 Figure 18.24 Graphic depicting hemorrhage within the thin inner dural border zone. A, Artist rendering of the normal meningeal dura and interface with the arachnoid mater. B, Hemorrhage at the DBCL shown in red. (From Mack J, Squier W, Eastman JT. Anatomy and development of the meninges: implications for subdural collections and CSF circulation. Pediatr Radiol. 2009;39:200–10, with kind permission from Springer Science and Business Media.)
Figure 18.24 Graphic depicting hemorrhage within the thin inner dural border zone. A, Artist rendering of the normal meningeal dura and interface with the arachnoid mater. B, Hemorrhage at the DBCL shown in red. (From Mack J, Squier W, Eastman JT. Anatomy and development of the meninges: implications for subdural collections and CSF circulation. Pediatr Radiol. 2009;39:200–10, with kind permission from Springer Science and Business Media.)When considering the neuropathology of SDH, it is important to remember that the DBCL is a delicate, thin (a few cell layers thick) microscopic layer, and when hemorrhage originates from the intradural venous plexus this delicate cell layer is disrupted. The claim by some that clinical SDH is actually contained within this veil-like DBCL is untenable as the DBCL is nonexistent in some locations, and where present, the thin cell layer would disrupt or disintegrate in the face of growing hemorrhage. In summary, there is no evidence to support a clinically relevant volume of subdural blood remaining confined to the fragile DBCL (pers. comm. with Lucy B. Rorke-Adams MD).
Whether the origin of SDH is from torn bridging veins, the inner DBCL, or both, trauma remains the most likely precipitating cause of SDH in the infant and young child. SDH should be considered an important marker of pathologic brain movement within the cranium (18, 77, 78, 81, 90, 103, 105, 162–164). Thus, for consistency and clarity the term subdural compartment, first popularized by Orlin and colleagues and more recently discussed by Mack, Squire, and Eastman is an acceptable term to describe the characteristic crescent-shaped hemorrhage that, when discovered in infants and young children, is commonly a result of trauma.
Other known and proposed causes of SDH in the neonate, infant, and young child that warrant further discussion include subdural compartment hemorrhage from childbirth and SDH observed in the setting of benign expanded SASs. Rare causes of nontraumatic SDH include: CSF volume depletion (e.g., following CSF diversion or shunting for hydrocephalus), coagulopathy, hematologic disorders, cerebral vascular malformations (e.g., AVM, arteriovenous fistula [AVF], cavernous malformation), and rare heritable and metabolic disorders (see Table 18.8).
Other hemic and nonhemic subdural collections
General imaging considerations
SDH is limited by dural barriers, including the dural venous sinus, falx cerebri and tentorium cerebelli. Location, size, and extent are variable (3, 9, 136). The false falx sign described by Osborn and Anderson, and observations made by Zimmerman and colleagues regarding the appearance of acute interhemispheric SDH, highlight for the radiologist the fact that in a younger patient, one should not normally detect the presence of the anterior inferior falx cerebri below the level of the inferior frontal horns (74, 75, 132, 165). Easily visualizing the anterior inferior “falx” should give the radiologist reason to suspect SDH (Fig. 18.25). Additionally, in the setting of known or suspected trauma, the observation of posterior fossa SDH, particularly near the foramen magnum should always be followed by a thorough search for craniocervical junction and/or brainstem/cervical cord injury (see Chapter 21) (74, 75, 132, 165).
 Figure 18.25 Acute interhemispheric SDH, the false falx sign. A, Axial NECT through the inferior aspect of the anterior cranial fossa in a four-year-old child who presented after a witnessed fall and closed head trauma shows linear increased attenuation (subdural compartment hemorrhage) within the inferior falx cerebri (arrow). The normal anterior inferior falx (below the level of the inferior frontal horns in an infant and young child) should be barely perceptible. B, Coronal reformation confirms hemorrhage within the inferior falx cerebri (arrow) superior to the dense skeletal crista galli.
Figure 18.25 Acute interhemispheric SDH, the false falx sign. A, Axial NECT through the inferior aspect of the anterior cranial fossa in a four-year-old child who presented after a witnessed fall and closed head trauma shows linear increased attenuation (subdural compartment hemorrhage) within the inferior falx cerebri (arrow). The normal anterior inferior falx (below the level of the inferior frontal horns in an infant and young child) should be barely perceptible. B, Coronal reformation confirms hemorrhage within the inferior falx cerebri (arrow) superior to the dense skeletal crista galli. The language used by radiologists to describe hemorrhagic and nonhemorrhagic subdural compartment collections is often confusing. Gaining consensus among radiologists on the use of terms describing subdural compartment hemorrhage and nonhemic collections, even within the same medical imaging department may be challenging. In addition to acute, subacute, and cSDH, the terms hematohygroma, hygroma, effusion, and empyema are used in clinical practice to describe specific subdural compartment collections (16, 22, 27–29).
Hematohygroma
The combination of SDH and CSF represents a hematohygroma. This admixture of CSF and hemorrhage typically occurs following trauma (45). In the hours to days following initial head trauma, serial cranial imaging will demonstrate progressive accumulation of CSF attenuation (CT) or signal intensity (MRI) fluid within the subdural compartment when the initial CT may have shown only a thin film SDH. A dependent sediment of acute hemorrhage (e.g., hematocrit effect) may be observed. Vinchon and colleagues emphasize that when estimating the age of the subdural hematohygroma, it is best to focus upon the sediment component when estimating age of hemorrhage (Fig. 18.26) (37, 120, 166).
 Figure 18.26 Hematohygroma following acute AHT. A 12-week-old infant presenting with seizures and obtundation. A, B, Sagittal and axial T1WI performed 72 hours after admission shows dependent hyperintense early subacute SDH (hematocrit effect) (arrows). Note the small right anterior middle cranial fossa lens-shaped EDH (arrowhead), probably venous in origin from injury to the sphenoparietal venous sinus. C, Axial T2WI shows layering of subacute SDH (arrows) with adjacent hyperintense supernatant (hematocrit effect). The right middle cranial fossa EDH exhibits features of early subacute hemorrhage (arrowhead). There were other features that supported that abuse was present and upon further investigation, the caretaker admitted to shaking the infant.
Figure 18.26 Hematohygroma following acute AHT. A 12-week-old infant presenting with seizures and obtundation. A, B, Sagittal and axial T1WI performed 72 hours after admission shows dependent hyperintense early subacute SDH (hematocrit effect) (arrows). Note the small right anterior middle cranial fossa lens-shaped EDH (arrowhead), probably venous in origin from injury to the sphenoparietal venous sinus. C, Axial T2WI shows layering of subacute SDH (arrows) with adjacent hyperintense supernatant (hematocrit effect). The right middle cranial fossa EDH exhibits features of early subacute hemorrhage (arrowhead). There were other features that supported that abuse was present and upon further investigation, the caretaker admitted to shaking the infant. Imaging considerations
Wells and Sty reported their CT observations among 55 children under the age of 3 years who suffered a single closed head trauma event (85% resulting from AHT). In 87% SDH was also detected on the initial NECT. In the week following trauma, serial NECTs demonstrated increasing CSF-like subdural accumulations in 80% (167). Their study and the work of Zouros and colleagues serve to remind radiologists that low attenuation (e.g., on NECT) nonhemic subdural collections can arise acutely and following a single traumatic event (45). There should not be a default assumption that the mixed attenuation (e.g., supernatant and sediment) subdural collection represents SDH of differing ages (37, 120, 166, 168, 169). Serial imaging of the hematohygroma will often demonstrate the originally detected SDH to become less conspicuous as the CSF attenuation (NECT) component grows (Fig. 18.27). The appropriate usage of the term post-traumatic subdural hygroma describes the situation where the initial head CT or MRI following head trauma shows no SDH or only a small CSF-like subdural fluid accumulation that enlarges over time (170).
 Figure 18.27 Hematohygroma following AHT. A 13-month-old infant found unresponsive in the crib. A, Axial NECT shows an acute/subacute left frontotemporal SDH (arrow) with mass effect. Note the low attenuation of the left cerebral hemisphere, indicative of edema. B, NECT repeated six hours after A, owing to posturing and seizures, shows the left SDH to be less apparent (arrow) and the presence of bi-frontal CSF-like subdural collections (arrowheads). C, Axial T2WI 24 hours after B, shows growing bi-frontal hyperintense, CSF-like subdural collections (arrows). The left posterior temporal SDH (arrowhead) is faintly apparent. D, Axial T2 FLAIR image demonstrates the frontal subdural collections to approximate the signal of ventricular CSF. E, Axial SWI shows CSF-like signal of the subdural collections (arrows) and small foci of hypointense SAH (arrowheads). The SS showed fractures in varied states of healing. Numerous facial and truncal bruises were observed.
Figure 18.27 Hematohygroma following AHT. A 13-month-old infant found unresponsive in the crib. A, Axial NECT shows an acute/subacute left frontotemporal SDH (arrow) with mass effect. Note the low attenuation of the left cerebral hemisphere, indicative of edema. B, NECT repeated six hours after A, owing to posturing and seizures, shows the left SDH to be less apparent (arrow) and the presence of bi-frontal CSF-like subdural collections (arrowheads). C, Axial T2WI 24 hours after B, shows growing bi-frontal hyperintense, CSF-like subdural collections (arrows). The left posterior temporal SDH (arrowhead) is faintly apparent. D, Axial T2 FLAIR image demonstrates the frontal subdural collections to approximate the signal of ventricular CSF. E, Axial SWI shows CSF-like signal of the subdural collections (arrows) and small foci of hypointense SAH (arrowheads). The SS showed fractures in varied states of healing. Numerous facial and truncal bruises were observed. Subdural hygroma
This author reserves using the term subdural hygroma SDHy to describe a CSF-like subdural compartment collection that appears following trauma. These nonhemic CSF-like collections may be detected within hours following trauma, or may slowly accumulate in the days following trauma (170–173).
The proposed mechanisms by which a hygroma forms following trauma is either by traumatic disruption of the arachnoid mater (e.g., flap valve mechanism), allowing CSF to accumulate in the subdural compartment, or from traumatic injury to the meninges and resultant vasomotor disturbance within the dura, inciting a cytokine-mediated response, thus, leading to CSF-like subdural fluid accumulation (as seen in Fig. 18.28) (170, 172, 174). Typically, the majority of post-traumatic SDHys will have resolved by three months following trauma. However, in a study by Ohno et al., more than half of the SDHys became cSDHs (175).
 Figure 18.28 Traumatic SDHy potential pathway to chronic subdural hemorrhage (cSDH).
Figure 18.28 Traumatic SDHy potential pathway to chronic subdural hemorrhage (cSDH). Imaging considerations
SDHys exhibit CT and MRI imaging features that are isoattenuating (CT) or isosignal (MRI) relative to CSF, or nearly so (Fig. 18.29). When subdural compartment fluid does not mirror CSF and bacterial meningitis is not a clinical consideration, then the differential diagnostic possibilities include: cSDH, hematohygroma, or the admixture of older and newer hemorrhage. There are clinical, chronologic, and imaging characteristics that aid in differentiating a SDHy from a cSDH (Table 18.5) (78).
 Figure 18.29 SDHy following accidental trauma. A three-year-old child following a witnessed fall from the playground monkey bars shows: A, NECT demonstrating small bi-frontal low attenuation subdural collections (arrows). B, Twelve hours later after worsening vomiting, the NECT demonstrates a growing right frontotemporal subdural CSF-like collection (arrow) and effacement of the left frontal SDHy. C, Coronal GRE image 36 hours following admission shows CSF-like fluid compatible with a right frontotemporal SDHy. All other MR pulse sequences showed the collection to be CSF-like. Note the small left convexity hygroma (arrow).
Figure 18.29 SDHy following accidental trauma. A three-year-old child following a witnessed fall from the playground monkey bars shows: A, NECT demonstrating small bi-frontal low attenuation subdural collections (arrows). B, Twelve hours later after worsening vomiting, the NECT demonstrates a growing right frontotemporal subdural CSF-like collection (arrow) and effacement of the left frontal SDHy. C, Coronal GRE image 36 hours following admission shows CSF-like fluid compatible with a right frontotemporal SDHy. All other MR pulse sequences showed the collection to be CSF-like. Note the small left convexity hygroma (arrow). | Feature | SDHy | cSDH | |
|---|---|---|---|
| Clinical: | Onset | Days to 3 weeks | Greater than 3 weeks |
| Head circumference | Static or decreasing | Increasing | |
| Imaging (CT/MRI): | Similar to CSF | Complex fluid | |
| Blood of varied ages | |||
| Lack of enhancing membrane | Enhancing neomembrane | ||
| Pathology: | Gross | Usually clear; +/- xanthochromic | Petroleum – like |
| Microscopic | Membranes uncommon | Membranes common | |
| Chemistry | CSF (increasing β total protein) | Blood products | |
Chronic SDH
The development of a cSDH in a previously healthy infant or young child following an acute traumatic SDH is uncommon. Most aSDHs following trauma resolve uneventfully (Fig. 18.30) (45, 78, 81, 171, 176).
 Figure 18.30 Formation of cSDH following aSDH.
Figure 18.30 Formation of cSDH following aSDH. Antecedent, hematohygroma, and/or hygroma represent underappreciated post-traumatic subdural compartment collections that many authors propose to be the most common pathway to the eventual formation of a cSDH (Fig. 18.28) (78, 81).
Following the formation of hygroma or hematohygroma, if the collection is slow to resolve or is large in size, the vascularization of the subdural neomembrane may lead to repetitive episodes of low pressure venous bleeding into the subdural compartment eventually forming a cSDH. Other patients at risk for developing repetitive venous hemorrhages within the low pressure subdural compartment include pediatric patients with shunted hydrocephalus, patients with post-traumatic encephalomalacia, or patients with a progressive neurodegenerative disorder (10, 177, 178).
Imaging considerations
For the radiologist, confidently distinguishing cSDH from hygroma is a common imaging challenge. The impact upon clinical management and potential forensic implications of accurately diagnosing cSDH versus hygroma cannot be overstated (see Table 18.5) (78, 179).
The presence of a membrane architecture within the SDH, enhancement of the endosteal and meningeal dura following intravenous contrast, loculated hemorrhage elements that do not settle dependently, and the attenuation and/or signal intensity features of the subdural fluid that are more complex than the CSF character of hygroma are all observations that aid in the diagnosis of a cSDH (Fig. 18.31) (45).
 Figure 18.31 Chronic and subacute SDH. An eight-month-old infant with seizures and bulging anterior fontanel. A, Axial NECT shows bilateral frontal low attenuation subdural collections (arrows). These show CT attenuation greater than ventricular CSF. High attenuation “membrane bound” hemorrhages are also seen in the frontal subdural compartments (arrowheads). The CT attenuation features are consistent with acute/subacute SDH. B, Thirty-six hours following the NECT, MRI was conducted and the axial T1WI shows the chronic SDHs (arrows) to be hypointense to gray matter and the “ante dependent” smaller bi-frontal SDHs are hyperintense (arrowheads). C, Axial T2WI demonstrates hyperintense chronic SDHs (arrows) and smaller loculated hypointense (early subacute) frontal SDHs (arrowheads). D, Axial T2 FLAIR image shows the chronic SDHs (arrows) to be slightly hyperintense to gray matter. Note the hyperintense bi-frontal subacute subdural hemorrhages (arrowheads). E, Coronal GRE image shows the hypointense membrane bound subacute hemorrhages (arrowheads). There were other signs supporting inflicted injury.
Figure 18.31 Chronic and subacute SDH. An eight-month-old infant with seizures and bulging anterior fontanel. A, Axial NECT shows bilateral frontal low attenuation subdural collections (arrows). These show CT attenuation greater than ventricular CSF. High attenuation “membrane bound” hemorrhages are also seen in the frontal subdural compartments (arrowheads). The CT attenuation features are consistent with acute/subacute SDH. B, Thirty-six hours following the NECT, MRI was conducted and the axial T1WI shows the chronic SDHs (arrows) to be hypointense to gray matter and the “ante dependent” smaller bi-frontal SDHs are hyperintense (arrowheads). C, Axial T2WI demonstrates hyperintense chronic SDHs (arrows) and smaller loculated hypointense (early subacute) frontal SDHs (arrowheads). D, Axial T2 FLAIR image shows the chronic SDHs (arrows) to be slightly hyperintense to gray matter. Note the hyperintense bi-frontal subacute subdural hemorrhages (arrowheads). E, Coronal GRE image shows the hypointense membrane bound subacute hemorrhages (arrowheads). There were other signs supporting inflicted injury. An additional challenge faced by the radiologist is distinguishing pre-existing cSDH in the setting of a mixed attenuation (CT) or mixed signal intensity (MRI) subdural compartment hemorrhage. When working through the imaging observations and interpretation of a complex SDH and when chronic components of hemorrhage are being considered in the differential diagnosis, IV MR contrast improves detection of membrane architecture within and marginating the SDH (Fig. 18.32) (22, 28).
 Figure 18.32 Chronic and aSDHs and internal subdural compartment membrane formation in the setting of AHT. A 10-month-old child presents to the ED in status, with bulging anterior fontanel, and facial and truncal bruising. A, Axial NECT shows a left frontal convexity and parafalcine mixed attenuation SDH that was interpreted as an acute hemorrhage (arrow). B, Twenty-four hours following the CT study, the MRI was performed. The coronal T2WI, shows a membrane bound medial left frontal hypointense SDH (aSDH component). There had been no settling of the hemorrhage in the 24 hours following the head CT. C, Coronal IV contrast-enhanced T1WI shows an enhancing lateral membrane (arrow) within the subdural compartment. Also note the small right parafalcine chronic SDH (curved arrow). In the author’s experience, the internal enhancing subdural compartment membranes are rarely detected with MRI prior to 2–4 weeks following initial SDH. (With kind permission from the Journal of the American Osteopathic College of Radiology. From Hedlund GL. Subdural hemorrhage in abusive head trauma: imaging challenges and controversies. J Am Osteopath Coll Radiol. 2012;1(1):23–30.)
Figure 18.32 Chronic and aSDHs and internal subdural compartment membrane formation in the setting of AHT. A 10-month-old child presents to the ED in status, with bulging anterior fontanel, and facial and truncal bruising. A, Axial NECT shows a left frontal convexity and parafalcine mixed attenuation SDH that was interpreted as an acute hemorrhage (arrow). B, Twenty-four hours following the CT study, the MRI was performed. The coronal T2WI, shows a membrane bound medial left frontal hypointense SDH (aSDH component). There had been no settling of the hemorrhage in the 24 hours following the head CT. C, Coronal IV contrast-enhanced T1WI shows an enhancing lateral membrane (arrow) within the subdural compartment. Also note the small right parafalcine chronic SDH (curved arrow). In the author’s experience, the internal enhancing subdural compartment membranes are rarely detected with MRI prior to 2–4 weeks following initial SDH. (With kind permission from the Journal of the American Osteopathic College of Radiology. From Hedlund GL. Subdural hemorrhage in abusive head trauma: imaging challenges and controversies. J Am Osteopath Coll Radiol. 2012;1(1):23–30.)In 1996, Kleinman and Ragland reported their experience using IV MR contrast to improve the conspicuity of thin convexity, basilar, and posterior fossa SDHs that were not readily appreciated on NECT owing to artifact from adjacent cortical bone (180). In recent years, multiplanar MRI, and particularly T2 FLAIR, has routinely improved detection of subtle SDHs (33, 37, 94, 181–183). It bears repeating that cSDHs on MRI may lack the expected GRE and/or SWI dephasing (e.g., hypointensity) (see Fig. 18.5).
Knowledge of the histologic features of the evolving SDH including histomorphologic changes provides the radiologist with insight into the age of the SDH. In their study of the histomorphologic alterations of 222 patients with SDH following closed head injury, Walter and colleagues detected the microscopic presence of reticular and collagen fibers (early membrane formation) within the SDH by day 10 and observed progressive membrane maturation within 20–30 days post injury (184). This pathomorphologic study supports this author’s personal neuroimaging experience over three decades indicating that membranes internal to SDHs are rarely detected with MRI less than 2–4 weeks following initial SDH and are more commonly observed at 4–6 weeks. Confident detection of membrane architecture within the SDH aids the radiologist in estimating the age of the SDH. MRI including IV contrast-enhanced T1WIs with fat saturation improves membrane detection within the SDH (Fig. 18.32).
Subdural effusion
Some radiologists use the terms effusion and hygroma synonymously. Others use the term subdural effusion to describe the proteinaceous subdural collection that may accumulate in the clinical setting of bacterial meningitis. Subdural effusion developing secondary to pyogenic meningitis may occur in pediatric patients who are responding to appropriate treatment for bacterial meningitis. These collections are not necessarily a complication of meningitis and most subdural effusions resolve uneventfully (6).
Imaging considerations
NECT shows increased attenuation of the subdural effusion compared to CSF. MRI offers improved sensitivity and specificity over NECT in the evaluation of subdural effusion, demonstrating T1 shortening (hyperintensity), T2 prolongation (hyperintensity), and T2 FLAIR hyperintensity compared to CSF. Diffusion-weighted images (DWI) including apparent diffusion coefficient (ADC) maps show increased diffusivity of the subdural effusion (Fig. 18.33).
 Figure 18.33 Subdural effusion secondary to Streptococcous pneumoniae meningitis. A, Axial NECT demonstrates bilateral low attenuation frontal subdural collections (arrowheads). The subdural fluid is of greater attenuation than frontal horn ventricular fluid. B, Axial T1WI following IV contrast shows hypointense frontal subdural effusions (arrowheads). Note the diffuse peripheral hyperintense leptomeningeal enhancement reflecting infection in the SASs. C, Axial DWI shows an absence of diffusion restriction within the frontal effusions (arrowheads). Restricted diffusion (hyperintensity on DWI) is compatible with subdural empyema.
Figure 18.33 Subdural effusion secondary to Streptococcous pneumoniae meningitis. A, Axial NECT demonstrates bilateral low attenuation frontal subdural collections (arrowheads). The subdural fluid is of greater attenuation than frontal horn ventricular fluid. B, Axial T1WI following IV contrast shows hypointense frontal subdural effusions (arrowheads). Note the diffuse peripheral hyperintense leptomeningeal enhancement reflecting infection in the SASs. C, Axial DWI shows an absence of diffusion restriction within the frontal effusions (arrowheads). Restricted diffusion (hyperintensity on DWI) is compatible with subdural empyema. The differential diagnosis for the NECT appearance includes cSDH, hygroma, and empyema (in the clinical setting of CNS infection) (16). With effusion and empyema a clear health history of pyogenic CNS infection is typically present.
Subdural empyema
Viscous infected subdural fluid represents an empyema. Subdural empyema most often develops as a complication of bacterial meningitis (e.g., infected subdural effusion) or an intracranial complication of sinusitis. Rarely, subdural empyema originates following bacterial sepsis, penetrating head trauma, craniotomy or craniectomy. Infection within the SAS (e.g., meningitis) associated with subdural effusion rarely leads to the development of subdural empyema. This fact speaks to the integrity and tight cellular junctions of the arachnoid barrier cell layer of the arachnoid membrane, thus confining disease within the SAS. The pediatric patient with subdural empyema clinically shows a lack of expected response to appropriate antibiotic therapy, often exhibiting persistent fevers, seizures, and CSF pleocytosis (6).
Imaging considerations
NECT shows increased attenuation of the subdural fluid when compared to CSF. A fluid debris level may be identified. Contrast-enhanced CT (CECT) or MRI appearance of empyema varies depending upon age of the infection, host immunologic response, and the infectious agent. Linear enhancement along the inner and outer margins of the subdural compartment collection is often seen. MRI helps to distinguish empyema from sterile proteinaceous subdural effusion by demonstrating diffusion restriction (e.g., DWI hyperintensity and hypointensity on ADC maps). These characteristics of diminished diffusivity are thought to be secondary to the viscous nature of the purulent infected subdural fluid (Fig. 18.34) (6, 16).
 Figure 18.34 Subdural empyema complicating sinusitis. A, Coronal T2WI shows a hyperintense left parafalcine and frontal convexity subdural compartment collection (arrows). Note the moderate mass effect and associated subfalcine herniation. B, Axial T1WI following IV contrast demonstrates inner and outer dural marginal enhancement of the extensive left parafalcine and lateral left frontal empyema (arrows). C, Axial DWI shows diffusion restriction (hyperintensity) within the empyema as a result of the viscous pus within the subdural compartment.
Figure 18.34 Subdural empyema complicating sinusitis. A, Coronal T2WI shows a hyperintense left parafalcine and frontal convexity subdural compartment collection (arrows). Note the moderate mass effect and associated subfalcine herniation. B, Axial T1WI following IV contrast demonstrates inner and outer dural marginal enhancement of the extensive left parafalcine and lateral left frontal empyema (arrows). C, Axial DWI shows diffusion restriction (hyperintensity) within the empyema as a result of the viscous pus within the subdural compartment. Intraventricular hemorrhage
Intraventicular hemorrhage (IVH) is the most common form of hemorrhage in preterm neonates. IVH following blunt pediatric head trauma is observed on NECT with a prevalence of approximately 3% (61, 84, 89, 185). Traumatic IVH often accompanies SDH and SAH. Isolated IVH following trauma is rare. The most common source of traumatic IVH is shearing of subependymal and choroidal veins. Other potential sources of IVH include retrograde reflux of SAH into the ventricles, dissection of hemorrhage from a periventricular parenchymal site (e.g., intra-axial hematoma, tumor, or AVM) and, rarely, hemorrhage originating from an aneurysm rupture (e.g., anterior communicating artery aneurysm rupture filling the anterior lateral ventricle).
Imaging considerations
IVH often layers dependently within the occipital horns. Other common locations of IVH include the third and fourth ventricles (Fig. 18.35). Hemorrhage that is predominantly basal in location should provoke a search for nontraumatic causes of hemorrhage (e.g., cerebral and spinal vascular malformations). Finally, isolated IVH is an uncommon finding in AHT (33, 80, 85, 89, 186, 187).
 Figure 18.35 Intraventricular hemorrhage, mixed signal intensity SDHs, and SAH in the setting of AHT. A, Axial NECT image demonstrates hyperattenuating IVH layering within the left occipital horn (arrow). Also note the high attenuation posterior interhemispheric, paratorcular and right lateral frontal SDHs (arrowheads) B, Axial T2 FLAIR image demonstrates a very subtle appearance of the IVH (arrow). C, Axial SWI shows the hypointense blooming of hemorrhage within the occipital horn (arrow). Also, note the dephasing (hypointensity) from SAH within the lateral cerebral (Sylvian) fissures (arrowheads). This two-month-old victim of inflicted injury had presented with new-onset focal seizures and also demonstrated healing rib and acromion fractures.
Figure 18.35 Intraventricular hemorrhage, mixed signal intensity SDHs, and SAH in the setting of AHT. A, Axial NECT image demonstrates hyperattenuating IVH layering within the left occipital horn (arrow). Also note the high attenuation posterior interhemispheric, paratorcular and right lateral frontal SDHs (arrowheads) B, Axial T2 FLAIR image demonstrates a very subtle appearance of the IVH (arrow). C, Axial SWI shows the hypointense blooming of hemorrhage within the occipital horn (arrow). Also, note the dephasing (hypointensity) from SAH within the lateral cerebral (Sylvian) fissures (arrowheads). This two-month-old victim of inflicted injury had presented with new-onset focal seizures and also demonstrated healing rib and acromion fractures. Subpial hemorrhage
The pia mater closely invests the brain and spinal cord following the normal cerebral convolutions, passing as a double layer into the cerebral sulci and fissures of the cerebellum.
Imaging considerations
On NECT and MRI, traumatic subpial hemorrhage is challenging to differentiate from a focal peripheral parenchymal hemorrhagic contusion and focal SAH. Subpial hemorrhage will hug the periphery of the brain, does not conform to a sulcus, and will not shift with patient positioning (Fig. 18.36). As with SAH, SDH, and IVH, it is important to fully appraise for associated comorbid cerebral, brainstem, and cerebellar injuries.
 Figure 18.36 Subpial hemorrhage following accidental closed head injury. A, Axial NECT shows a peripheral right frontal peri-pterional high attenuation punctate hemorrhage (arrow) which was coup to impact. B, Twenty-four hours later, MRI was conducted and the axial T1WI shows a peripheral hyperintense hemorrhagic focus (arrow). C, Axial T2WI demonstrates hypointense hemorrhage (arrow). Note that the hemorrhage does not conform to a sulcus, as would be expected with SAH. D, Axial SWI shows focal blooming or hypointensity of the subpial hemorrhage (arrow). Also note the right temporal parietal cephalohematoma (the site of head impact).
Figure 18.36 Subpial hemorrhage following accidental closed head injury. A, Axial NECT shows a peripheral right frontal peri-pterional high attenuation punctate hemorrhage (arrow) which was coup to impact. B, Twenty-four hours later, MRI was conducted and the axial T1WI shows a peripheral hyperintense hemorrhagic focus (arrow). C, Axial T2WI demonstrates hypointense hemorrhage (arrow). Note that the hemorrhage does not conform to a sulcus, as would be expected with SAH. D, Axial SWI shows focal blooming or hypointensity of the subpial hemorrhage (arrow). Also note the right temporal parietal cephalohematoma (the site of head impact). Challenges in the interpretation of SDH and nonhemic subdural fluid collections
Childbirth and SDH
Birth-related SDH is common following vaginal delivery and even after uncomplicated cesarean section. It has been proposed that stretching of intracranial structures such as the falx cerebri, tentorium cerebelli, and cortical and bridging veins during childbirth results in SDH (188). As the sensitivity and specificity of imaging technology has improved, it is not surprising that the reported prevalence of asymptomatic birth-related SDH has increased (189–192).
Understanding the typical appearance and the natural evolution of these parturitional hemorrhages has important clinical care and forensic implications, as there are “experts” who opine that birth-related subdural bleeding may precisely mimic the intracranial hemorrhage patterns of AHT in the infant (189, 190, 193–195). To clarify the natural evolution of birth-related hemorrhage, Rooks and colleagues prospectively studied and reported the prevalence and evolution of intracranial hemorrhage in 101 asymptomatic term newborns. Seventy-nine (78%) were born via vaginal delivery; the remainder required vacuum assistance, forceps, or cesarean section. They evaluated the incidence, distribution, size, and appearance of the SDH. All patients were evaluated with MRI (1.5 Tesla). They reported a 46% incidence of SDH among their cohort. All patients with SDH received serial MRI examinations until the SDHs had resolved (191). A year earlier, in a study of vaginally delivered neonates imaged at 3.0 Tesla, Looney et al. reported a birth-related SDH incidence of 26% (196).
In the study by Rooks et al. the SDHs were predominantly detected posterior in location (posterior hemispheric fissure, parafalcine, paratentorial, falcotentorial, and adjacent to the occipital poles). All 46% of the patients with SDH had a supratentorial SDH component; 43% of these patients also had posterior fossa SDH. Most SDHs in their patients were thin (<3.4 mm), and 93.8% showed resolution by 1 month of life. All SDHs had resolved by three months. One patient with an initially detected thin SDH, when re-imaged at 26 days showed a left frontal SDH that was reported as “spontaneous.” Rooks and colleagues concluded their prospective study stating, “SDH after 1 month of age is unlikely to be birth related” (191). Whitby and colleagues reported similar temporal resolution findings of birth-related SDH among nine neonates who on MRI showed resolution of SDH by four weeks (197).
Re-bleeding from a birth-related SDH is very unlikely to explain SDH detected in infants beyond 1–2 months of age, particularly those with clinical neurologic deterioration (see Figs. 18.43, 18.45) (78, 81, 194).
Imaging considerations
Detection of SDH in the acute stage (<3 days of age) with MRI is challenging, as T1WI and T2WI signal intensity is similar to the cerebral and cerebellar cortex in this stage. GRE imaging and SWI show hemorrhage dephasing (hypointensity) in the acute stage, thus improving hemorrhage detection (Fig. 18.37) (22, 26–28, 191, 193, 197). To summarize, birth-related SDH is typically thin, posteriorly located, above and below the tentorium and most have resolved by one month.
 Figure 18.37 Birth-related SDH in a term newborn. At four days of age, MRI was performed to investigate apnea in this product of a normal vaginal delivery. A, Axial T1WI shows bilateral thin hyperintense occipital SDHs (arrows). B, Axial T2WI shows that these hemorrhages are hypointense (consistent with early subacute hemorrhage). C, Coronal GRE image demonstrates the value of a susceptibility sequence to show the presence of a thin hypointense left tentorial SDH (arrow). GRE and SWI sequences are particularly helpful for detecting acute birth-related SDH that may be nearly isointense to brain cortex on T1- and T2-weighted sequences. (Used with kind permission from the Journal of the American Osteopathic College of Radiology. From Hedlund GL. Subdural hemorrhage in abusive head trauma: imaging challenges and controversies. J Am Osteopath Coll Radiol. 2012;1(1):23–30.)
Figure 18.37 Birth-related SDH in a term newborn. At four days of age, MRI was performed to investigate apnea in this product of a normal vaginal delivery. A, Axial T1WI shows bilateral thin hyperintense occipital SDHs (arrows). B, Axial T2WI shows that these hemorrhages are hypointense (consistent with early subacute hemorrhage). C, Coronal GRE image demonstrates the value of a susceptibility sequence to show the presence of a thin hypointense left tentorial SDH (arrow). GRE and SWI sequences are particularly helpful for detecting acute birth-related SDH that may be nearly isointense to brain cortex on T1- and T2-weighted sequences. (Used with kind permission from the Journal of the American Osteopathic College of Radiology. From Hedlund GL. Subdural hemorrhage in abusive head trauma: imaging challenges and controversies. J Am Osteopath Coll Radiol. 2012;1(1):23–30.)BESSI and SDH
In a busy medical imaging department, it is common to encounter infants with macrocrania and benign enlargement of the subarachnoid spaces in infancy (BESSI). Numerous terms have been used to describe this reversible condition including: benign extra-axial collections of infancy, benign macrocephaly of infancy, benign external hydrocephalus (BEH), external hydrocephalus, and arachnoidomegaly (6, 168, 199). The key word in the description of this entity is benign. Benign enlargement of the SASs in infants is a clear description of this transient entity with a good prognosis and is currently in widespread use (6).
These infants typically come to clinical attention as a result of rapid head growth or for the evaluation of macrocrania. They are otherwise neurologically normal, achieving age-appropriate developmental milestones. In most cases there is a family history of macrocephaly. Males show greater representation than females. Parents may offer a history that they also had large heads in infancy. A common clinical scenario is a newborn with a high normal head circumference (HC) at birth and increasing HC in the first few months of life. By early to mid infancy, the HC rises above the 95th percentile. The natural history of BESSI is stabilization of the HC by roughly 18 months of age and subsequent normalization of HC and SASs between 24 and 36 months of age (200, 201).
It is hypothesized that the normalization of the subarachnoid fluid volumes around the second year of life coincides with establishment of an upright posture, closure of the anterior fontanel, and an accompanying drop in intracranial venous pressure, thus providing a maturational stimulus to the arachnoid villi. Furthermore, age-related developmental immaturity of the arachnoid villi, and impaired CSF absorption at the level of the nasal mucosal lymphatic pathway underlying the cribriform plate have been implicated in the development of benign expanded SASs (6, 202). In support of this latter theory is the fact that infants and young children who experience elevated intracranial venous pressure (e.g., extracorporeal membrane oxygenation [ECMO] therapy) demonstrate a similar pattern of expanded SASs (202). When an imaging study is ordered, it is typically performed to address concerns over possible pathologic causes of macrocrania (e.g., hydrocephalus or occult SDH). Hypomagnesemia and the amplification of the physiologic imbalance between skull and brain growth are less commonly appreciated possible contributors to reversible hydrocephalus or BESSI (203).
Infants with BESSI demonstrate expanded SASs over the cerebral convexities, particularly bi-frontally and along the anterior interhemispheric fissure. Prominent CSF containing SASs within the sylvian and basal cisterns, and mild ventricular dilation are typical findings. Cortical sulci are conspicuous and mildly prominent, and the subarachnoid fluid volumes are balanced over the cerebral convexities showing a symmetric interface with the falx cerebri. The SAS fluid echogenicity (ultrasonography [US]), attenuation (CT), and signal intensity (MRI) should precisely follow the imaging characteristics of CSF (204).
A thoughtful differential diagnosis for the cause of expanded SASs should be considered prior to a default diagnosis of BESSI. This is particularly true when there are clinical signs and symptoms, and imaging findings that militate against the diagnosis of BESSI. The differential diagnosis of enlarged SASs includes: dehydration, malnutrition, total parenteral nutrition, adrenocorticotropic hormone (ACTH) and corticosteroid use, chemotherapy, meningitis, and neoplastic pachymeningitis (leukemia, leptomeningeal PNET, melanoma, and metastatic disease). Enlargement of the SASs can be an early harbinger of a genetic disorder (mucopolysaccharidosis, achondroplasia, and agenesis of the corpus callosum) (78, 203). Bilateral cerebral atrophy will also lead to enlarged SASs. Additionally, and perhaps most importantly, is the recognition that thin film SDHs may impede normal CSF absorption and, thus, lead to an increased accumulation of subarachnoid fluid. The interpreting radiologist must be alert to this subtle yet very important observation, recognizing that the presence of SDHs may antedate the development of expanded CSF containing SASs (78).
Enlargement of the SASs has been theorized as a risk factor for the development of SDH (200, 201, 205–215). Furthermore, some physicians posit and testify in court that infants with BESSI are clearly at a greater risk for the development of SDH either spontaneously or following minimal trauma that may precisely mimic the SDHs observed in cases of AHT (168, 193, 207, 208, 210, 216). The proponents of this hypothesis opine that in the setting of expanded SAS fluid, movement of the brain exerts tension on cortical and bridging veins, thus leading to venous tearing and SDH. A biomechanical model proposed by Papasian and colleagues suggests that when the width of the SAS increases to 6 mm (compared with the normal 3–4 mm) the traversing cortical dural veins are at risk of tearing with translational brain movement (209). A counterpoint to this view is the finite element (FEM) model proposed by Raul and colleagues asserting that prominent subarachnoid fluid spaces may actually have a stabilizing effect upon bridging veins and thus be protective against the development of SDH (217).
In an early observational study by Hamza et al., 13 infants with BESSI were followed for 30 months. NECT was used serially to evaluate the extra-axial spaces. None of the 13 developed SDH (201). McKeag and colleagues evaluated 177 patients less than 2 years of age with macrocrania who had been diagnosed with enlargement of the SASs with either MRI or CT. SDHs were detected in 4 patients (2.3%). All four patients with SDH underwent evaluations for child abuse that included consultation with the CPT, skeletal survey (SS), and an ophthalmologic examination. One of the four children with SDH had additional injuries (e.g., rib fractures) (218). Greiner et al. investigated the prevalence of subdural collections in children less than two years of age presenting with macrocrania. A cohort of 108 patients with BESSI who had been imaged with CT and/or MRI revealed subdural collections in 6 patients. Two patients were identified as being victims of abuse and their subdural collections were large and complex (septated/hemorrhagic). The four remaining patients showed subdural collections that were small, homogeneous, and nonhemorrhagic (219). These clinical cohort studies emphasize that SDHs in the context of macrocrania and expanded subarachnoid CSF spaces are uncommon and warrant a thoughtful evaluation for trauma, including inflicted injury (7, 27, 81, 168, 220).
What is known from clinical practice is that normal infants and young children have larger SASs than do older children and adults. The diagnosis of BESSI should be reserved for developmentally normal infants and young children (3 months to ∼2 years) and where imaging studies characterize the extra-axial fluid to be of CSF quality and to reside within the SAS. The benign fluid designation must be questioned when macrocrania is accompanied by delayed development, and encephalopathy. Macrocrania and extra-axial fluid accumulation in the first two months of life, and fluid in the subdural compartment do not represent the features of BESSI (221).
Despite the fact that minor household trauma is common in ambulatory infants and young children, SDHs resulting from these events are distinctly uncommon. Therefore, the view that subdural bleeding commonly occurs spontaneously or with minor head trauma in the setting of BESSI should be viewed with caution; additional prospective studies with serial imaging are needed to resolve the issue. Currently, at the author’s hospital, infants and young children who are discovered to have expanded subarachnoid fluid spaces and coexistent SDH undergo a thorough evaluation by the CPT, since some of these patients will be found to have suffered AHT (126).
Imaging considerations
Cranial sonography with color Doppler, NECT including coronal and sagittal reformations, and multiplanar MRI (particularly SWI and T2 FLAIR sequences), are particularly helpful in assigning the location and characterizing the nature of the extra-axial fluid when BESSI is suspected. The infant with BESSI will typically exhibit a skull-to-cerebral cortex distance greater than 5 mm. Although consensus is lacking on the limits of enlargement of the SASs, the defined upper limits of normal for cranial–cortical width has been reported to range between 3 and 10 mm. The normal distance from the lateral wall of the superior sagittal venous sinus to the surface of the cerebral cortex (sinocortical width) is 2–10 mm (203). Visualization of cortical venous structures traversing the extra-axial fluid, symmetric fluid interface with the falx cerebri, prominence of the sulci, and CSF imaging characteristics all point to CSF within the SAS (Fig. 18.38) (6, 7, 22, 202, 220). The cortical vein sign, as proposed by McCluney et al., decribes the “positive cortical vein sign” when the expanded extra-axial fluid space exhibits features of an enlarged SAS compartment and not subdural collections which would compress the SAS and the veins running through it (veins displaced inwardly against the brain surface) (155).
 Figure 18.38 BESSI. A, Axial NECT shows normal prominent frontotemporal subarachnoid fluid spaces in an infant with macrocephaly and normal developmental milestones. Note the mild enlargement of the frontal horns and the isoattenuating SAS CSF compared to the ventricular fluid. B, Coronal T2WI demonstrates the normal cortical venous vessels traversing the SASs (arrows) (cortical vein sign). Subdural fluid collections would generally displace these vessels inwardly against the brain surface. C, Coronal image from a cranial sonogram with color Doppler shows a normal large left parafalcine cortical vein (orange vessel) traversing the expanded CSF containing SAS in an infant with macrocephaly.
Figure 18.38 BESSI. A, Axial NECT shows normal prominent frontotemporal subarachnoid fluid spaces in an infant with macrocephaly and normal developmental milestones. Note the mild enlargement of the frontal horns and the isoattenuating SAS CSF compared to the ventricular fluid. B, Coronal T2WI demonstrates the normal cortical venous vessels traversing the SASs (arrows) (cortical vein sign). Subdural fluid collections would generally displace these vessels inwardly against the brain surface. C, Coronal image from a cranial sonogram with color Doppler shows a normal large left parafalcine cortical vein (orange vessel) traversing the expanded CSF containing SAS in an infant with macrocephaly. Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


