

Abstract
First principles approaches to the protein structure prediction problem must search through an enormous conformational space to identify low-energy, near-native structures. In this paper, we describe the formulation of the tertiary structure prediction problem as a nonlinear constrained minimization problem, where the goal is to minimize the energy of a protein conformation subject to constraints on torsion angles and interatomic distances. The core of the proposed algorithm is a hybrid global optimization method that combines the benefits of the αBB deterministic global optimization approach with conformational space annealing. These global optimization techniques employ a local minimization strategy that combines torsion angle dynamics and rotamer optimization to identify and improve the selection of initial conformations and then applies a sequential quadratic programming approach to further minimize the energy of the protein conformations subject to constraints. The proposed algorithm demonstrates the ability to identify both lower energy protein structures, as well as larger ensembles of low-energy conformations.
Keywords: Protein tertiary structure prediction, Hybrid global optimization algorithm
1 Introduction
The protein folding problem can be simply stated: Of all the possible conformations, how does a denatured protein fold into its unique structure? Leventhal’s paradox raised the concern of how a protein could find the native functional state if the time scale for folding is only on the order of milliseconds or even microseconds [1]. The basic premise of protein folding is Anfinsen’s thermodynamic hypothesis, which states that the native structure of a protein can be determined directly from its sequence by identifying the global minimum free energy conformation [2]. However, the major difficulty in this approach is efficiently distinguishing the global minimum energy confirmation from the numerous local minima of the free energy landscape. As Levinthal’s paradox suggests, a random search through all possible protein conformations is unreasonable, so the careful application of global optimization methods has been useful in addressing these problems.
Due to the complexity of simulating the protein folding problem, comparative modeling techniques have been developed to exploit the information contained in proteins with experimentally-determined structures. Comparative modeling techniques require the identification of a suitable template from a homologous protein and the alignment of the target protein to that template. Once an appropriate template and alignment have been selected, a model of the target in the structurally conserved regions is built, the positions of the side chain atoms and loop residues are added, and a refinement and/or evaluation of the target structure. Comparative modeling techniques have traditionally been based on sequence-sequence alignment methods such as BLAST [3], but can be significantly improved using profile-sequence comparisons [4], hidden Markov models [5], or profile-profile alignment methods [6].
Fold recognition and threading methods have also been proposed to detect remotely homologous proteins for comparative modeling efforts [7–9]. The premise of fold recognition strategies is the idea that protein folds are more conserved than protein sequences and therefore the number of protein folds is orders of magnitudes less than the number of protein sequences [10, 11]. Threading methods, a well-studied class of protein fold recognition algorithms, attempt to fit or “thread” the target protein sequence onto the template protein structure. Several methods have been proposed to address the protein threading problem, including dynamic programming techniques [12], iterative approaches [13, 14], and optimization-based approaches [15, 16].
The majority of the remaining approaches to the tertiary structure prediction problem are based on the premise that the minimum energy conformation of a protein corresponds to its native state. These approaches can be roughly divided into two categories: (1) methods that depend on homology-based information for their searches (i.e., statistical potentials and/or biased conformational searches) and (2) methods that are independent of homology-based information (i.e., physics-based potentials and conformational searches with optimization algorithms).
One popular homology-based tertiary structure prediction approach, fragment assembly, uses short subsequences of known structures from the Protein Data Bank [17] to build an overall protein structure. This structure can then be optimized using scoring functions or statistical potentials in combination with optimization algorithms. Baker and co-workers have used simulated annealing approaches to assemble compact structures from these protein fragments [18, 19]. These compact structures can then be optimized through the minimization of a scoring function, based on conformational statistics of known proteins, with Monte Carlo minimization techniques.
Many of the more successful protein structure prediction approaches are hybrid methods that combine comparative modeling techniques with the minimization of a scoring function. One example of this is the work of Skolnick and co-workers, which combines multiple sequence comparisons, threading, a united atom lattice model, the optimization of a scoring function, and clustering for the prediction of protein structures [20–22]. Their methods have been applied iteratively to the ab initio modeling of small proteins with promising results [23]. Other tertiary structure prediction algorithms focus on the minimization of a knowledge-based scoring function, using a lattice-based approach [24] or Monte Carlo techniques [25].
Methods that are independent of homology information make direct use of Anfinsen’s thermodynamic hypothesis [2], exploring the conformational space of a protein in an attempt to identify the minimum free energy structure of the protein in its environment. These physics-based approaches are typically computationally demanding, requiring times on the order of CPU years on a single processor for all but the smallest proteins. Despite this heavy demand, physics-based protein folding studies have several advantages over homology based approaches. These advantages include the ability to (1) predict structures independent of the existence of homologous structures, (2) extend predictions to proteins in different environmental conditions, and (3) provide insight into the mechanism, thermodynamics, and kinetics of protein folding.
Monte Carlo and molecular dynamics techniques have been used for a number of protein folding studies. Dill and co-workers proposed a zipping and assembly model using replica exchange molecular dynamics for the folding of small, single-domain proteins [26]. A hierarchical approach using a Metropolis Monte Carlo algorithm has been applied to protein structure prediction (LINUS) by identifying conformational biases from a discrete set of moves and evaluating a simplified physics-based force field [27, 28]. Pande and co-workers have folded the protein villin to an RMSD of 3 Å using molecular dynamics implemented through their popular distributed grid computing application, Folding@Home [29].
Scheraga and co-workers introduced a simplified united-residue force field (UN-RES) to the problem of protein structure for their initial calculations and then refine the coarse protein structure models using an all-atom potential [30–32]. In this united-residue force field, the representation of each amino acid residue is reduced to two interaction sites. This reduction allows the stochastic conformational space annealing algorithm to more efficiently identify low energy regions of the conformational space [33–35]. The use of the united-residue force field with replica-exchange Monte Carlo and minimization search techniques was also investigated [36].
One successful prediction method is the first principles ASTRO-FOLD protein folding approach developed by Floudas and coworkers [37]. The main thrusts of this approach are (1) α-helical prediction through detailed free energy calculations [38], (2) a mixed-integer linear optimization formulation for the β-sheet prediction [39], (3) derivation of secondary structure restraints and loop modeling, and (4) the application of hybrid global optimization algorithms to tertiary structure prediction. The goal of the ASTRO-FOLD tertiary structure prediction approach is to minimize the potential energy of a protein subject to constraints imposed on the dihedral angles and distances. The algorithm is a combination of the deterministic αBB global optimization algorithm, a stochastic global optimization algorithm, and a molecular dynamics approach in torsion angle space [37, 40].
The use of the αBB global optimization algorithm guarantees convergence to the global minimum solution by a convergence of upper and lower bounds on the potential energy minimum. Upper bounds to this model can be obtained through local minimizations of the original non-convex problem. The addition of separable quadratic terms to the objective and constraint functions produces a convex lower bounding function. With these bounding functions, the problem can be iteratively branched over the variable space, fathoming portions when a region’s lower bound rises above the best upper bound. The highly nonlinear force field and vast conformational space present an exceptionally difficult problem for deterministic approaches by themselves. The introduction of torsion-angle dynamics methods to quickly identify feasible low energy conformers and a stochastic optimization method, conformational space annealing, for enhanced searching of the upper bounding function can greatly increase the power of this algorithmic approach. These hybrid algorithms, their convergence properties, and their parallel implementations have been discussed in detail [41, 42]. The ASTRO-FOLD framework can be applied to proteins of any size, but the detailed energetics and deterministic guarantees of the approach are best-suited for small to medium-sized proteins (up to approximately 200 amino acids in length). The methodology has been applied to a varied set of proteins throughout this range [37] and in a recent double blind prediction [43]. The proposed hybrid global optimization algorithm is an extension of the tertiary structure prediction work of Floudas and co-workers [37, 40–42]. The goal of this proposed method is to combine effective global optimization techniques with efficient local minimization strategies.
Due to the broad scope of the protein structure prediction literature, the methods presented here are merely a representative sample of the algorithmic variety that exists. A more thorough description of protein structure prediction methods is available in a number of recent reviews [44–47].
2 Methods
The proposed tertiary structure prediction algorithm will be presented in the following order. In Sect. 2.1, a suitable potential energy function is selected for the tertiary structure prediction problem. In Sect. 2.2, techniques for introducing bounds on the torsion angles and distances within a protein are discussed. Given an appropriate energy function and a set of constraints, Sect. 2.3 describes the mathematical formulation of the protein structure prediction problem as a minimization problem. The use of sequential quadratic programming techniques as a local minimization tool to address the constrained nonlinear programming problem of protein structure prediction is discussed in Sect. 2.4. The application of constrained local minimization techniques is heavily dependent upon the identification of initial feasible points with low energy. Sect. 2.5 describes how a torsion angle dynamics algorithm can be used to satisfy both the dihedral angle and distance constraints and also avoid large numbers of steric clashes between atoms. Further improvement of the initial feasible points can be achieved using the rotamer optimization techniques in Sect. 2.6 that optimize the placement of the side-chain atoms using a discrete library and a fixed protein backbone. Sect. 2.7 describes the application of the αBB deterministic global optimization algorithm to problem of protein structure prediction, incorporating the algorithmic components of Sects. 2.4–2.6. The conformational space annealing algorithm, a combination of simulated annealing and genetic algorithms, is a stochastic optimization technique that can be applied to the protein structure prediction problem as described in Sect. 2.8. The αBB and CSA algorithms can be effectively combined into a hybrid algorithm to retain the advantages of each individual method. This hybrid algorithm and its parallel design are outlined in Sect. 2.9. A formal presentation of the algorithms is available as Supplementary Material and can be accessed at http://titan.princeton.edu/ProtAlg2009/.
2.1 Potential energy function
The importance of the protein structure prediction problem has led to the development of a wide variety of force fields. Physics-based force fields include energetic contributions of atomic bonds, angles, and torsion angles, as well as non-bonded interactions such as van der Waals interactions and electrostatics. AMBER [48] and CHARMM [49] are physics-based atomic potentials of this type. When the atomic bond lengths and bond angles are fixed to constant values, the energetic contributions of these terms can be ignored. The ECEPP [50], ECEPP/3 [51] and ECEPP-05 [52] force fields fix these values to allow protein conformations to be represented by only its torsion angles.
The ECEPP/3 atomistic level force field was chosen as the energy function. The ability to represent the protein conformation using only the torsion angles is a significant advantage for minimization approaches as it drastically reduces the variable space that must be considered. This force field calculates the potential energy of the protein as the sum of electrostatic, non-bonded, hydrogen bonded and torsional contributions. Equation (1) details the form of each of these terms.
| (1) |
The interatomic distance of the atomic pair (ij), rij, is present in the first three terms. qi and qj are dipole parameters for the respective atoms, where the dielectric constant of 2 has been incorporated. Fij assumes a value of 0.5 for 1–4 interactions and 1.0 for 1–5 and higher interactions. The non-bonded parameters specific to the atomic pair, Aij and Cij are also necessary to calculate the non-bonded contributions. Likewise, and Bij are the hydrogen bonded parameters specific to the atomic pair. The sets ES, NB, HB are the atomic pairs (ij) that have electrostatic, non-bonded and hydrogen bonding interactions, respectively. The set TOR are the torsion angles, k, that contribute to the torsion potential contributions.
2.2 Constraints
The tertiary structure prediction problem for proteins requires the search of a vast conformational space. Various constraints on both torsion angles and interatomic distances can be imposed to narrow the size of this conformational space. One source of constraints is through the identification and use of homologous proteins with structures that have been determined by experimental techniques [53]. An alternative approach to generating constraints is to approach the protein folding problem using a framework-based algorithm. A logical way to generate constraints for such a problem, such as the Astro-Fold framework [37], is to first predict the protein secondary structure using either homology-based techniques or free energy modeling, then to predict the arrangement/topology of the secondary structure elements [39, 54], and finally to predict the loop regions of the protein [55, 56].
There are a variety of ways to constrain the dihedral angles based on the prediction of secondary structure elements. The ideal, right-handed α-helix adopts a φ angle of approximately −57° and a ψ angle of approximately −47° [57]. In a folded natural protein, the geometry of an α-helix may deviate from these values depending upon its environment. As shown in Table 1, Klepeis and Floudas have used two similar sets of dihedral angle bounds for residues predicted to be part of an α-helix.
Table 1.
Dihedral angle bounds for α-helix residues
The ideal antiparallel β-strand adopts a φ angle of approximately −139° and a ψ angle of approximately 135°, whereas the parallel β-strand has a φ angle of −119° and a ψ angle of 113° [57]. The geometry of the β-strands also deviates from these values in folded proteins depending on the environment and other factors. As shown in Table 2, Klepeis and Floudas have used two similar sets of dihedral angle bounds for residues predicted to be part of a β-strand. These values are used regardless of the existence of a parallel or an antiparallel orientation.
Table 2.
Dihedral angle bounds for β-strand residues
Distance constraints can also be introduced based on predicted secondary structure elements or predicted topology. The formation of an α-helix is dependent upon the formation of a hydrogen bond between the carbonyl oxygen at residue i and the backbone – NH at position i + 4. Klepeis and Floudas [37] introduced bounds on the distances between the Cα atoms of these helical residues to enforce the formation of the hydrogen bonding network. Knowledge of β-sheet topology can also be used to introduce bounds on the distances before the application of the tertiary structure prediction algorithm. Klepeis and Floudas [37] used bounds on the distances between the Cα atoms for β-sheets. These bounds are non-local and help to enforce the formation of a β-sheet between two opposing β-strands. The lower and upper distances used for these bounds are presented in Table 3.
Table 3.
Bounds on distances based secondary structure
| Secondary structure type | dL | dU |
|---|---|---|
| α-helix residue (i, i + 4) | 5.5 | 6.5 |
| β-sheet contact | 4.5 | 6.5 |
2.3 Formulating as a minimization problem
Given the definition of an energy function in Sect. 2.1 and a set of constraints in Sect. 2.2, the protein structure prediction problem can be formulated as a minimization problem. This formulation has been described in detail previously [40], so only the key aspects of formulating the problem will be covered here.
The prediction of protein tertiary structure can be formulated as either a constrained or unconstrained minimization problem. The unconstrained formulation requires the minimization of a hybrid energy objective function that combines both the ECEPP/3 potential energy as well as penalty values for the violation of the enforced dihedral angle and distance bounds as shown in (2). In this equation, θ is the vector of dihedral angles representing the protein conformation, EECEPP/3 (θ) is the ECEPP/3 potential energy contribution of this conformer, Eres (θ) is an energy term quantifying the restraint violations, and Wres is a weight factor. The weight factor should be large enough such that when this objective function is minimized, the sum of the bound violations is driven to zero.
| (2) |
The energy term quantifying the restraint violations includes contributions from both the dihedral angle bounds and the distance bounds. The violations of the distance bounds are quantified in a quadratic form using a simple square well potential. Equations (3)–(4) illustrate the separation of the distance bound violations into lower and upper bounding terms, respectively. The distance violation terms are individually weighted by the values and , which only contribute when the calculated distance dj assumes a value below the lower bounding distance or exceeds the upper bounding distance , respectively. A similar form of a quadratic function for violations of the dihedral angle bounds using a square well potential can be derived for unconstrained minimization, but becomes more complicated due to the periodic nature of dihedral angles [40].
| (3) |
| (4) |
An alternative to the formulation of (2) is to address the dihedral angle and distance bounds as mathematical constraints and minimize an objective function that contains only the potential energy contribution. Equation (5) describes the formulation of the protein tertiary structure prediction problem as a constrained minimization problem. The distance bound violations may be grouped as a single constraint, or separated into any number of subsets of multiple constraints, l = 1, …, Ncon, as a specific problem may require. The distance bound violation value of a given conformation θ for the set of bounds l is represented by . This violation is required to be less than or equal to a reference parameter , which may be zero or some small value to account for uncertainties in the values of and . The dihedral angle values, θk are also constrained to be within lower ( ) and upper ( ) bounding values. If no information is available to constrain a dihedral angle θk, the default range of [−π, π] is used.
| (5) |
2.4 Local minimization
Nonlinear minimization problems with nonlinear constraints are typically represented in the form of (6). The objective function, f(x) is a nonlinear function to be minimized subject to the constraints r(x), where x represents the vector of variables that can be altered to find the minimum value of f(x). It should be clear that the problem presented in Sect. 2.3 can be cast in this fashion.
| (6) |
Constrained nonlinear minimization techniques can be addressed using sequential quadratic programming (SQP) approaches, augmented Lagrangian methods, reduced-gradient methods, and interior-point methods.
The overall structure of sequential quadratic programming approaches solve the nonlinear programming problem of (6) by using both major and minor iterations. The major iterations identify a series of line search steps that converge to a solution, x*, that satisfies first order conditions for optimality. These iterates are defined by a step length, α, and a search direction, p, which are combined to move from the current point x̄ to the next point x. The search direction can be identified by solving the quadratic program of (7). In this equation, g(x) is the gradient vector of first order derivatives of f(x), H is a positive definite quasi-Newton approximation to the Hessian of the Lagrangian, and J(x) is the Jacobian matrix of first derivatives of r(x).
| (7) |
The formulation in (7) is solved using a second iterative procedure, which constitute the minor iterations of the overall SQP algorithm. Once the search direction p, has been identified, an appropriate step length is calculated that decreases the value of a merit function. This merit function measures the quality of each iterate by weighing both the current value of the function to be minimized, f(x̄), and the feasibility of the solution x̄ with respect to any non-linear constraints. Further details of the SQP method are available elsewhere [58–61].
Although the basics concepts of a constrained nonlinear programming method remain constant across implementations, there may be considerable differences between various solvers in both applicability and performance. The NPSOL package [61] is especially attractive for the protein structure prediction minimization problem of Sect. 2.3 as it requires relatively few evaluations of the computationally expensive ECEPP/3 potential energy function.
One important parameter that is varied during the minimizations using NPSOL is the line search tolerance. This parameter can assume a value in the range of [0.0, 1.0), with a default value of 0.9. As the minimization takes a step, α, during a given iteration, the line search parameter controls the accuracy of the step when compared to the minimum of the merit function. For applications of the protein structure prediction problem, the use of the NPSOL algorithm with a single line search tolerance converges to an infeasible point that cannot be improved for as many as 50% of the minimizations in complex problems with hard to satisfy distance constraints. By performing several minimizations and altering the value of the line search tolerance, the algorithm can effectively and robustly identify low-energy, feasible protein conformations in the region of the initial conformation.
The overall approach for each constrained nonlinear minimization to be performed as part of the proposed tertiary structure prediction algorithm in this paper is outlined below and presented formally in the Supplementary Material. The main idea of this approach is to perform repeated calls of NPSOL using varying values of the line search tolerance until a termination criteria is met. The minimization of the given conformer terminates if a specified number of initial point improvements have been achieved or a thorough exploration of the range of line search parameters has been explored.
Receive and store the initial point which is the vector of dihedral angles representing the protein conformation to be minimized
Initialize the upper bounding value of the distance constraint violations
Store the initial point as the best solution obtained so far
Initialize the number of initial point improvements to zero
Initialize the number of local minimizations to zero
Initialize the line search tolerance to 0.9
-
While the number of initial point improvements is less than 2 and the number of local minimizations is less than 8:
Reset the current initial point to the initial point specified in (1)
Call the NPSOL constrained local minimization routine with an iteration limit of 8000 steps
if the minimized protein conformation satisfies the distance constraint violation and has a lower energy than the best protein conformation, save this new conformation as the best conformer and increment the number of improvements (Strategy: Retain the lowest energy)
if the minimized protein conformation does not satisfy the distance constraint violation and the violation value of this conformation is less than the violation value of the best protein conformation, save this new conformation as the best conformer and increment the number of improvements (Strategy: Retain the conformation that is the closest to feasibility)
Increment the number of local minimizations
Decrement the line search tolerance by 0.1
Return the best protein conformation identified by this minimization routine
The application of constrained local minimization techniques is heavily dependent upon the identification of initial feasible points with low energy. Methods for identifying feasible initial conformations are presented in Sect. 2.5. The energy values of these initial conformations can be effectively minimized using the rotamer optimization techniques described in Sect. 2.6. By applying the constrained local minimization routines to these improved initial conformations, the time required by the local minimization is significantly reduced and the energetic quality of the resulting conformers is noticeably improved.
2.5 Initial point (conformation) selection
The identification of initial feasible points is critical to the success and efficiency of the constrained nonlinear minimization algorithm. The quality of an initial conformation can be measured by how well the conformation satisfies the imposed distance and dihedral angle constraints and how well the conformation avoids steric clashes that result in large energetic penalties.
If the exact distance values are available for all pairs of atoms, the Cartesian coordinates for these atoms can be determined uniquely (with the exception of translational and rotational degrees of freedom and inversion) using metric matrix distance geometry [62–64]. For protein tertiary structure prediction problems, the distance values possess neither the exactness nor the completeness required for the unique embedding of a single set of atomic coordinates. Distance geometry algorithms such as EMBED [64] and dgsol [65] can be used to produce molecular conformations that satisfy a set of sparse distance constraints.
Several alternatives to distance geometry algorithms exist, including molecular dynamics in Cartesian space [66, 67], variable target function methods [68, 69], and molecular dynamics in torsion angle space (torsion angle dynamics) [70–72]. The various algorithms for protein structure determination are reviewed in further detail elsewhere [73].
The basic torsion angle dynamics routine in the initial implementation of the ASTRO-FOLD framework has been replaced by a more detailed torsion angle dynamics annealing procedure by interfacing with the CYANA (Version 2.1) software package [71]. The number of structures that satisfy both the distance and dihedral angle constraints after this routine is significantly increased, especially in the cases of distance constraints with tight bounds or large numbers of distance constraints. The algorithmic steps are outlined below and presented formally in the Supplementary Material.
Initialize the protein sequence, the bounds on the torsion angles, and the lower and upper interatomic distance bounds. The protein sequence and distance bounds remain constant throughout the algorithm, so they must be initialized only once. The torsion angle bounds vary based on the current αBB subregion and therefore must be initialized for the first conformer of each subproblem.
Initialize the weight value for van der Waals lower limit constraints, wvdw, to 0.5; the weight value for torsion angle constraints, waco, to 104; and the weight value for lower and upper distance constraints, wdco, to 5.0. The large value for waco is used to ensure the torsion angle bounds are satisfied at the completion of the annealing routine.
Create a random initial protein conformation
Reduce the atomic radii of heavy atoms and hydrogen atoms to focus the initial stages of the annealing routine on satisfying the provided distance and torsion angle bounds
Minimize the initial protein structure with 100 steps of conjugate gradient minimization including constraints up to levels 3, 10, 25, 50, 100, and 150. The effects of a distance constraint or van der Waals constraint is only included when the number of residues separating the two interacting atoms is less than the specified level value.
Perform the first round of TAD for Nsteps/3 starting at the highest temperature value, Thigh and decreasing as a function of the number of steps completed, s
Increase the atomic radii of the heavy atoms to their actual values
Minimize the current protein structure with 50 steps of conjugate gradient minimization including all constraints
Perform the second round of TAD for Nsteps/3
Increase the atomic radii of the hydrogen atoms to their actual values
Increase the weight value for van der Waals lower limit constraints, wvdw, to 1.0
Minimize the current protein structure with 50 steps of conjugate gradient minimization including all constraints
Perform the third round of TAD for Nsteps/3
Increase the weight value for van der Waals lower limit constraints, wvdw, to 2.0
Minimize the current protein structure with 50 steps of conjugate gradient minimization including all constraints
Perform the final round of TAD for 200 steps at the final temperature, Tfinal
Minimize the current protein structure with 1000 steps of conjugate gradient minimization including all constraints
2.6 Rotamer optimization
The interactions of the side chain atoms in a protein are crucial to both the stability and specificity of its native state. The placement of these side chain atoms can be posed as a combinatorial problem by approximating the continuous space of atom placements using a library of rotational isomers. The rotational isomers, commonly referred to as rotamers, are residue-specific and are typically represented as a set of dihedral angles, χ1…N, where N is the number of side chain dihedral angles required to specify the placement of this side chain.
A variety of rotamer libraries have been proposed to represent the likely conformations of side-chain atoms in native protein structures. One of the earliest rotamer libraries was built from merely 19 well-refined proteins and contained only 67 rotamers [74]. Dunbrack and Karplus included the dependence of a rotamer conformation on the local backbone conformation of a protein in their library, producing the first backbone-dependent rotamer library [75]. Lovell et al. created a “Penultimate” rotamer library by applying stringent criteria to select only highly resolved and refined protein structures and to eliminate specific side-chains with high B-factors, atomic clashes, or uncertain ring orientations [76]. A thorough review of the history, limitations and role of rotamer libraries can be found in [77].
Many algorithms have been introduced to address the problem of protein side-chain prediction using rotamer optimization techniques and these approaches can be divided into two categories. The first class of algorithms are able to guarantee convergence to the placement of rotamers that yields the minimum energy. The Dead-End-Elimination (DEE) approach was one of the earliest optimization approaches that has been applied to the side-chain placement problem [78]. By proving that a better alternative rotamer positioning exists, DEE eliminates single rotamers or rotamer combinations that cannot be part of the minimum energy solution. These DEE methods have improved to be used alone for rotamer optimization [79], or in concert with other search algorithms, such as an A* search [80] or a residue-rotamer-reduction approach [81]. Alternative methods with optimality guarantees for the side-chain placement problem include mixed-integer linear programming [82, 83] and graph theory approaches [84].
As an alternative to algorithms that provide guaranteed convergence to the global minimum conformation, a number of heuristic algorithms have been proposed that are faster than many of the previously described algorithms. These approaches include the use of Monte Carlo searches [85, 86], cyclical search methods [75, 87], genetic algorithms [88], and mean field optimization [89]. Desmet et al. proposed the Fast and Accurate Side-Chain Topology and Energy Refinement (FASTER) approach that combines the DEE techniques with a multi-pass algorithm that systematically overcomes local minima of increasing order [90].
Rotamer optimization is an integral part of many protein structure prediction approaches. Many of the earliest ab initio protein structure prediction methods separate the problem into two distinct subproblems: (a) the generation of a protein backbone structure and (b) the assignment of side-chain atom positions [91]. Rotamer optimization techniques continue to be important in homology modeling methods, fold recognition and threading approaches, fragment assembly algorithms, and ab initio protein structure prediction [77, 92].
A rotamer optimization stage has been introduced in the proposed tertiary structure prediction algorithmic framework prior to the constrained nonlinear minimization of a protein conformation. Given an initial protein backbone, either from torsion angle dynamics runs or through conformational space annealing, the goal of the rotamer optimization stage is to remove any steric clashes that may exist between protein side chains and provide a better starting point for local minimization. In this respect, rotamer optimization acts as an efficient local minimizer. Therefore, the selected approaches have the following goals (a) they are fast, heuristic-based approaches and (b) they search a large rotamer library.
The efficiency of the rotamer optimization algorithms outlined here can be significantly enhanced by the use of an approximate energy function. This approximate energy function will be used to closely represent the repulsive energetic penalty that results from steric clashes between the atoms in the Lennard-Jones and hydrogen bonding potential energy contributions in the ECEPP/3 force field. Both the torsion and electrostatic potential energy terms are ignored in this approximation because they are heavily outweighed by the energy of repulsion. Any interaction energy between two atoms that is less than 2 kcal/mol is ignored. The contribution of repulsion is then approximated by a piece-wise linear function that intersects the interaction energy at values of 2, 5, 10, 20, 50, 100 kcal/mol. The linear approximation between 50 and 100 kcal/mol is extended for distances that lead to interaction energy values beyond 100 kcal/mol.
A number of rotamer optimization algorithms, especially those that are combinatorial in nature, function by dividing the energy contribution into two parts, the self energy and the pair energy. The self energy of rotamer k at residue position i, , represents the energetic interaction of this rotamer with all atoms that remain fixed during the rotamer optimization. Thus, the moveable side chain atoms of residue i are evaluated against all the backbone atoms, the Cβ side chain atoms, and any other immovable side chain atoms, such as Hα or Hβ, to obtain the rotamer self energy. The pair energy, , is evaluated for the placement of rotamer k at residue position i and rotamer l at residue position j. This energy contribution evaluates the interactions of the moveable side-chain atoms at both residue positions concurrently. Equations (8)–(9) define the self and pair energies, where Ri is defined as the set of rotamers that are valid for the amino acid at position i, the indices m, n represent atoms on the protein, Mi,k is the set of movable atoms for residue i (whose positions are defined by rotamer k), and Fi is the set of fixed atoms for residue i.
| (8) |
| (9) |
Given this separation of the energy terms, the rotamer optimization problem is concisely stated as (10).
| (10) |
The rotamer optimization strategy for the proposed tertiary structure prediction approach is divided into three stages. The first rotamer optimization algorithm is an application of the FASTER algorithm. The original FASTER algorithm has been shown to produce nearly identical results to DEE-based rotamer optimization methods but is nearly 100–1000 times faster [90]. In their computational studies, the initialization stage (specifically the calculation of the self and pair energy contributions) actually becomes the dominant and rate-limiting computational phase. In this application, the FASTER algorithm is applied to the current protein structure using the reasonably-sized Penultimate rotamer library. The algorithmic details can be found in [90]. For tertiary structure prediction problems with distance constraints for the moveable side chain atoms (such as those encountered with NMR structure prediction and refinement problems), the self and pair energies evaluated for the FASTER approach are penalized (with a constant penalty term) if the rotamer assignment would result in a violation of these distance constraints.
The second rotamer optimization algorithm described here is a simple cyclical search algorithm. The algorithm steps through the entire Xiang and Honig rotamer library at each residue position, saving changes that lead to an improvement in the ECEPP/3 energy function. The approximate energy function is used to accelerate the search, by only visiting the detailed energy function if the approximation suggests the rotamer positioning in question may be in the same energetic range. The algorithmic steps are outlined below and presented formally in the Supplementary Material.
All backbone atoms are inserted into an atomic grid, which will be used to identify possible neighbors for the frequent computation of the approximate energy function.
Save the coordinates of the original protein conformer as the first rotamer choice. Subsequently, retrieve and store the coordinates of the possible rotamers using the Xiang and Honig rotamer library (specifically the library generated with 297 protein structures, 96% coverage, and a 10° torsion angle tolerance) as the remaining rotamer choices.
-
Randomly rearrange the order of the residues to be visited by the rotamer optimization algorithm. For each residue i,
Remove the side-chain atoms of residue i from the atomic grid
The energy of the approximate energy function for each rotamer k is calculated as the sum of three contributions. First, the energy contributions from the intra-residue atom pairs that are connected by three bonds are calculated. Second, the energy of all intra-residue pairs connected by more than three bonds is added to the energy contribution. Finally, a query to the atomic grid is made for each side-chain atom to determine possible inter-residue interactions that may contribute to the energy. Each of these possible interactions is considered and added to rotamer energy where appropriate.
The ECEPP/3 energy is calculated for the original rotamer at residue i
Set the approximate energy cutoff to the approximate energy of the original plus a tolerance
for each rotamer k, if the approximate energy is below the cutoff value, calculate the ECEPP/3 energy for this rotamer. If this energy is lower than the original (or previous rotamer update) energy at residue i AND the selection of this rotamer does not increase the violation of any imposed distance constraints, update the rotamer selection and approximate energy cutoff for residue i.
if a rotamer change has been made in step (3e), update the χ angles and rotamer coordinates of residue i
Insert the side-chain atoms of the new (or original, if unchanged) rotamer k for residue i into the atomic grid
While the iteration limit has not been reached, go to step (3).
The final rotamer optimization algorithm presented here is a random local search algorithm that proceeds in a similar fashion to the cyclical search algorithm. Instead of using the Xiang and Honig rotamer library, random rotamers are generated from a narrow Gaussian distribution in the region of the current rotamer. This algorithm is intended to provide a degree of refinement after the application of either or both of the first two algorithms, helping to bridge the gap to the continuous χ angle space while retaining some of the favorable properties of discrete rotamer searches. The algorithmic steps are outlined below and presented formally in the Supplementary Material.
All backbone atoms are inserted into an atomic grid, which will be used to identify possible neighbors for the frequent computation of the approximate energy function.
Save the coordinates of the original protein conformer as the first rotamer choice. Subsequently, generate and store the coordinates of the possible rotamers by generating random χ angles from a Gaussian distribution with a mean value of the original χ angles and a standard deviation of 10°. For the current implementation, 50 random rotamers are generated for each residue i.
Randomly rearrange the order of the residues to be visited by the rotamer optimization algorithm. For each residue i, follow steps (3a)–(3f) of the cyclical search algorithm.
While the iteration limit has not been reached, go to step (2).
The use of the rotamer optimization as a local minimizer provides a better initial protein conformation for subsequent constrained local minimizations with solvers such as NPSOL. Any reasonable rotamer optimization protocol for this framework should require significantly less CPU time than the gradient based minimization approach. For rotamer optimizations of the protein PDB:1o2f, the associated CPU times are approximately 1 second for the FASTER approach, 8 seconds per cyclic search iteration, and 1 second per random local search iteration. The total time of approximately 25 seconds for the rotamer optimization protocol compares favorably to constrained local minimization runs that require on the order of 5–15 minutes per protein conformation.
The overall approach to the problem of rotamer optimization for the tertiary structure prediction algorithm can be summarized as (1) a single application of the FASTER algorithm using the Penultimate rotamer library, (2) two iterations of a cyclic search algorithm using a large rotamer library from Xiang and Honig’s research efforts and (3) ten iterations of a random local search algorithm using randomly generated χ angles in the vicinity of the original rotamer position. This approach demonstrates the ability to act as an efficient local minimizer and can provide better initial configurations to the NPSOL constrained local minimization.
2.7 αBB deterministic global optimization
The αBB algorithm is a deterministic global optimization approach that can provide theoretical guarantees of convergence to the global optimal solution of a wide-variety of optimization problems with twice-continuously differentiable objective and constraint functions [93–97]. The algorithm achieves convergence by creating a nondecreasing series of lower bounds on the global optimum as well as a non-increasing series of upper bounds on this optimum. These two series of bounds eventually converge to the global optimum value of the optimization problem. The αBB approach has been previously applied to the protein structure prediction problem [37, 40].
The use of the αBB global optimization algorithm guarantees the identification of the global minimum solution by a convergence of upper and lower bounds on the potential energy minimum. The upper bound on the global minimum is obtained by constrained nonlinear local minimization on any protein structure. The lower bound is determined by creating a valid convex underestimating function and identifying its minimum function value. The algorithm converges by successively partitioning regions of conformational space at every level of a branch and bound tree. The lower bounding formulation can be written as shown in (11).
| (11) |
The term LECEPP/3 (θ) is the lower bounding function of the force field on the current region and can be expressed by (12). The αθi terms are nonnegative convexification parameters that can be calculated rigorously through a variety of approaches and must be greater than −1/2 of the minimum eigenvalue of the Hessian of the energy function over the domain in question [98].
| (12) |
The term is the convex relaxation of the distance bound violation term, , as shown in (13). This relaxation produces a convex overestimation of the feasible region as defined by the constraint on the distance bound violations.
| (13) |
Given the solutions of the lower and upper bounding problems, the algorithm proceeds by branching on a region of dihedral angle space. Bisecting a single dihedral angle across all nodes at a given level of the tree has been suggested as an appropriate branching strategy for protein conformation problems [40]. The subproblem with the infimum of all the minimum values of the lower bounding functions is identified as the next candidate for branching to ensure non-decreasing lower bounds. The upper bounding values of a region can be identified using constrained nonlinear local minimization techniques, such as those found in Sect. 2.4. A non-increasing sequence of these upper bounds can be determined by selecting the current upper bound as the minimum value of all previously determined protein conformations. Any subregion where the lower bounding value exceeds the current upper bound can be fathomed, as it can no longer contain the minimum of the potential energy function.
The implementation of the αBB algorithm will be presented as three phases, (i) initialization, (ii) algorithm control and (iii) single iteration. This separation will be useful in Sect. 2.9, where a parallel implementation of a hybrid global optimization algorithm is proposed. The initialization phase must be executed prior to the other two phases to identify global variables, dihedral angle bounds, distance bounds, α values, and other necessary information.
Select the set of dihedral angles that will function as the global variables. Typically the backbone φ and ψ dihedral angles are chosen due to their influence on the overall structure of the protein. Unlike the global variables, the remaining dihedral angles are local variables and are not used as branching variables.
Input lower and upper bounds on all dihedral angle variables. Methods for identifying these bounds are presented in Sect. 2.2.
Input distance bounds to be used as additional constraints. These bounds may be from secondary structure location or topology prediction or other sources. In the implementation described here, all of the distance bounds are combined into a single constraint equation
Input the value of for the right-hand side of the constraint equation. This value is typically 0 (strictly enforcing distance bounds), but can be loosened if a large number of tight distance bounds are introduced or if other information suggests uncertainty in these bounds.
Identify the initial α values used to produce convex underestimators of the energy and constraint functions.
Initialize the upper bounding value to an arbitrary large positive value.
The αBB algorithm control handles the iterative nature of the method. The lower and upper bounding values of each subregion are stored here and used to identify the non-increasing sequence of upper bounds and non-decreasing sequence of lower bounds. In addition, this information is used to identify branching directions and candidates for fathoming. The control of the algorithm proceeds as shown below and is presented formally in the Supplementary Material.
If this is not the first iteration, select the subproblem with the lowest lower bound value that remains on the queue of subproblems. If this is the first iteration, select the full problem that represents the entire space of valid dihedral angles.
Perform a single αBB iteration on the selected problem as specified below.
If the lower bound of the subproblem is greater than the best upper bound, fathom the subproblem and go to step (7).
If the upper bound of the subproblem is less than the best upper bound, store the new best upper bound.
Partition the current subproblem along one of the global variables defined in the initialization phase and add both of the new subproblems to the queue of remaining subproblems.
Update the iteration count.
If the queue of remaining subproblems is not empty, proceed to step (1).
A single αBB iteration focuses on identifying the lower and upper bounding function values for a given subregion. For the protein structure prediction applications, torsion angle dynamics and rotamer optimization methods are included as part of the algorithm structure. The torsion angle dynamics approach, as described in Sect. 2.5, allows for the rapid identification of protein conformations that are feasible with respect to the distance and angle bound constraints. The rotamer optimization methods, as described in Sect. 2.6, function as quick and effective local minimizers. The integration of these two components into an αBB iteration and the overall layout of this iteration is described below and presented formally in the Supplementary Material.
Define the dihedral angle bounds and distance bounds that will be applied to the protein conformations in the current subregion
Define a threshold number of TAD structures that could be used in the constrained nonlinear minimization operations. This number will be the sum of the number of lower bounding minimizations and number of upper bounding minimizations to be performed.
-
While a minimum number of TAD runs have not been executed OR (the number of structures satisfying the violation threshold is not sufficient AND the maximum number of TAD runs have not been executed), do the following:
Execute the CYANA TAD annealing schedule described in Sect. 2.5.
Retrieve the dihedral angle values representing the conformer generated by this application of TAD.
Adjust the dihedral angles to the nearest acceptable value if they do not satisfy the initially specified bounds.
Evaluate the energy function, the lower bounding function, and the sum of the distance constraint violations.
if the sum of the distance constraint violations satisfies a violation threshold value, store the new conformer and increment the variable representing the number of structures satisfying this threshold.
Given the ensemble of conformers generated by the TAD algorithm, identify a distance constraint violation cutoff value for structures that could be used in the constrained nonlinear minimization operations.
Perform rotamer optimization according to the method outlined in Sect. 2.6 on the conformers that satisfy this violation cutoff.
Re-evaluate the energy function and the lower bounding function.
-
While the number of lower bounding minimizations has not been performed (theoretically only one minimization is necessary for the convex lower bounding function, but more may be required to due to practical limitations in the solver or other considerations):
Select the conformer that has the minimum distance constraint violation value. If two or more structures have the same distance constraint violation value, select the structure with the minimum lower bounding function value.
Remove this conformer from the list of conformers eligible for lower bounding minimizations.
Perform constrained nonlinear minimization of the lower bounding function according to the procedure outlined in Sect. 2.4.
Do 10 more iterations of the random local search rotamer optimization algorithm described in Sect. 2.6.
Re-evaluate the energy function and the lower bounding function.
Store the minimized conformer.
If the conformer with the lowest lower bounding function value exceeds the best upper bounding function value previously achieved, this subregion can be fathomed and the algorithm proceeds to the next iteration. If not, then the algorithm proceeds.
-
While the number of upper bounding minimizations has not been performed:
Select the conformer that has the minimum distance constraint violation value from the results of the TAD runs as well as the lower bounding minimizations. If two or more structures have the same distance constraint violation value, select the structure with the minimum lower bounding function value.
Remove this conformer from the list of conformers eligible for upper bounding minimizations.
Perform constrained nonlinear minimization of the upper bounding function according to the procedure outlined in Sect. 2.4.
Do 10 more iterations of the random local search rotamer optimization algorithm described in Sect. 2.6.
Re-evaluate the energy function and the lower bounding function.
Store the minimized conformer.
If the lower bounding function value of this conformer is less than the previously identified values, store the new lower bound for this subregion.
Return the lower bound for the subregion and the ensemble of conformers produced by the upper bounding minimizations.
2.8 Conformational space annealing
The conformational space annealing (CSA) algorithm has been proposed by Scheraga and co-workers as a stochastic optimization technique that is well-suited for the problem of protein structure prediction [33–35, 99–101]. This approach can be classified as a member of the simulated annealing class of algorithms [102], where the conformational search space is initially unrestrained but becomes gradually narrowed as the algorithm progresses. This slow reduction of the achievable conformations ideally restricts the search to the lowest energy regions where the global minimum conformation is likely to occur. The CSA algorithm combines this simulated annealing approach with elements of a genetic algorithm as a hybrid stochastic global optimization approach. Unlike the αBB deterministic global optimization approach presented in Sect. 2.7, this stochastic optimization technique offers no theoretical guarantee of finding the global minimum protein conformation in finite time.
The outline of the basic CSA approach used as part of the proposed protein tertiary structure prediction algorithm is presented below and also presented formally in the Supplementary Material. The source of the protein conformers to create the initial bank and add additional conformers to the bank will be the solutions of the αBB subproblems, which will be described in detail in Sect. 2.9. The three genetic operations of steps (2(a)ii), (2(a)iii), and (2(a)iv) are similar to the genetic operations previously implemented by Scheraga and co-workers [33, 100].
Receive and store the set of 20 protein conformations to be used as the initial bank
-
While the maximum number of conformational space annealing iterations has not been completed:
-
While the number of iterations since the last bank update is less than the bank update frequency:
Randomly select a protein conformation from the bank
Randomly “mutate” between 1 and 4 φ, ψ, χ1 dihedral angles to a value from another protein conformation in the initial bank. Subject this altered conformer to rotamer optimization and local minimization as described below. If the minimized conformation satisfies the bank acceptance criteria, update the bank of protein conformers (4 times)
Randomly select a second conformation from the current bank. Replace a randomly selected continuous range of 1/8 of the total number of dihedral angles in the first conformation with the values from the second conformation. This is known as a “crossover” operation. This altered conformer is then subject to rotamer optimization and local minimization as described below. If the minimized conformation satisfies the bank acceptance criteria, update the bank of protein conformers (3 times)
Perform a crossover operation similar to step (2(a)iii), but replace 1/4 of the total number of dihedral angles in the first conformation with the values from the second conformation. Subject this altered conformer to rotamer optimization and local minimization as described below. If the minimized conformation satisfies the bank acceptance criteria, update the bank of protein conformers (3 times)
Increment the number of iterations since the last bank update
Increment the total number of conformational space annealing iterations
Receive a set of 10 protein conformations to add to the bank of conformers
Reset the number of iterations since the last bank update
-
The bank acceptance criteria is dependent upon a measure of distance between two protein conformers. The distance between two protein conformers, i and j, in conformational space is defined as the absolute deviation of the differences of the dihedral angle values as shown in (14). In this equation, and represent the dihedral angle k for the protein conformations i and j, respectively.
| (14) |
Given the definition of a distance metric in (14), it is straightforward to define an average pairwise distance value over all of the protein conformations in the CSA bank. If Nbank is the current size of the CSA bank, the average pairwise separation distance between conformations in the bank, Davg is defined by (15). It is obvious that this equation is only meaningful for Nbank ≥ 2.
| (15) |
A cutoff distance, Dcut, is defined to prohibit the protein bank from becoming heavily biased towards one region of the conformational space during the early stages of the CSA algorithm. This cutoff distance is initialized to Davg/2. Since this algorithm is an annealing method, the value of Dcut should be gradually reduced as the algorithm progresses. Several annealing schedules have been proposed to reduce Dcut exponentially [41, 100], linearly [42], or as a function of the difference between the lower bounding function value (from the αBB results) and potential energy value of the conformers in the CSA bank [41]. The annealing schedule used for the proposed tertiary structure prediction algorithm relates the value of Dcut to a weighted combination of the Davg value and the improvement/convergence of the αBB upper and lower bounds as shown in (16). In this equation, δαBB is the current difference between the αBB lower and upper bounds, which is compared to the initial difference of these bounds, δαBB,0
| (16) |
Given the definition and annealing schedule of this Dcut value, the following procedure describes how an altered and minimized conformer is evaluated for acceptance into the conformational bank of the CSA algorithm.
Calculate the distance, D, between the proposed conformer and each of the other conformers in the CSA bank according to (14). Store the conformer i that has the minimum distance to the proposed conformer.
If the distance between the proposed conformer and the nearest conformer i is less than or equal to Dcut, then the proposed conformer replaces conformer i in the bank if and only if the proposed conformer has a lower energy value than conformer i.
If the distance between the proposed conformer and the nearest conformer i is greater than Dcut and the proposed conformer has a lower energy than at least one of the current bank conformers, then the proposed conformer replaces the current bank conformer with the highest energy value.
The local minimization strategy for the trial conformations of the CSA approach is outlined below and presented formally in the Supplementary Material. The use of rotamer optimization algorithms, in a similar approach to the one described in Sect. 2.7, provides a good starting point for further minimization by eliminating the large energetic penalties that accompany any steric clashes between side chain atoms. The altered conformer with improved side-chain atom positions can then be subject to constrained nonlinear minimization techniques where the positions of all the protein atoms (represented by the dihedral angles) are allowed to vary.
Define the dihedral angle bounds and distance bounds that will be applied to the protein conformations in the current subregion. This is typically the original set of dihedral angle and distance bounds for the protein system
Receive a protein conformation that has been altered by the previously described operations
Perform rotamer optimization according to the method outlined in Sect. 2.6 on the altered protein conformer.
Re-evaluate the energy function and the distance constraint violation value
Perform constrained nonlinear minimization of the energy function according to the procedure outlined in Sect. 2.4.
Do 10 more iterations of the random local search rotamer optimization algorithm described in Sect. 2.6.
Re-evaluate the energy function and the lower bounding function.
Return the minimized conformer.
2.9 Hybrid algorithm and parallel implementation
The deterministic nature of the αBB provides a theoretical guarantee of convergence to the global optimal solution. Due to the lower-bounding problems that must be solved over each problem domain, the computational requirements of this algorithm are significant. The stochastic CSA algorithm cannot provide any information regarding the lower bound of the energy function within a given region of conformational space, but it is an efficient method of identifying and improving protein conformations that place an upper bound on the potential energy. The ideal algorithm would be a combination of these two techniques, which would yield an algorithm that can both identify lower bounds on protein conformers and efficiently identify and refine protein conformations with low energies.
Two parallel hybrid algorithms have been previously proposed to combine the benefits of the αBB and CSA algorithms. The first approach, an integrated hybrid method, executes the αBB and CSA algorithms in succession as the overall minimization approach [41]. In this approach, an αBB iteration is performed to identify a low-energy conformation, which is passed to the CSA approach as a trial conformation and subjected to mutation and crossover operations. The second approach, the alternating hybrid algorithm, is based on the separation of the αBB and CSA algorithms [42]. The αBB algorithm is repeatedly performed to build and update the bank of conformers necessary for CSA approach. These conformers are then subject to the standard CSA mutation and crossover operations followed by local minimization. The alternating hybrid approach is used as the basic framework for the algorithm proposed in this article for tertiary structure prediction.
Several modifications of the parallel implementation of the alternating hybrid algorithm have also been made. The duties of the two control processors have been combined and assigned to a single overall control processor. This combination is effective because the work distributed to the work processors requires several orders of magnitude more time for processing than for the communication associated with delivering the work and receiving the results. Very little delay of the work distribution due to communication was observed. The initial implementation of the algorithm assigned a fixed set of processors to specific duties (αBB iterations, CSA iterations). Since no CSA work exists until a bank has been filled by low energy structures from the αBB algorithm, these processors would sit idle during the first rounds of the algorithm. This inefficiency has been removed by initially assigning all processors to perform iterations of the αBB method. Once the initial CSA bank of conformers is filled, the control processor sends a message to a subset of the work processors indicating that their duties have changed. The algorithmic steps of the improved hybrid global optimization approach are shown below and presented formally in the Supplementary Material.
Initialize the primary processor with control of N secondary processors, each with an initially unassigned duty.
Load the protein sequence, the initial dihedral angle bounds, and the lower and upper distance bounds.
Artificially branch k times to create enough initial problems to assign one per secondary processor (2k ≥ N). Initialize the lower bounding value of each subproblem to −∞
Send out the initial problems to the N secondary processors.
Place any remaining initial subproblems on a priority queue of αBB problems, sorted such that the subregion with the infimum of the lower bounding values receives the highest priority
-
While an iteration limit or time limit has not been reached:
Probe for a secondary processor that is waiting to return results.
-
If the secondary processor has completed αBB work:
Receive the lower bounding function value for the subregion and a list of the low energy conformations identified during the αBB search.
Write the low energy conformations to a file.
Add the low energy conformations to a queue of αBB solutions.
If the current conformations have a lower energy than the best identified energy, store the new minimum energy.
If the subregion is not a candidate for fathoming, branch on the next global variable dimension and add the two new subproblems to the priority queue of αBB problems. The lower bounding value of these new subproblems is initialized to the lower bounding value of the original subproblem
-
If the duty of this secondary processor is unassigned:
If the requisite number of processors have already been assigned to CSA duties, assign αBB duty to the secondary processor. Remove the highest priority subproblem from the priority queue of αBB problems and send it to this secondary processor.
If the requisite number of processors have not already been assigned to CSA duties, assign CSA duty to the secondary processor. If the initial CSA bank is not established, add this processor to a stack of CSA secondary processors waiting for work. If the initial CSA bank can now be established due to the size of the queue of αBB solutions, send out the initial CSA work to all of the processors on stack. If the initial CSA bank was already established, send out the next unit of CSA work to the current secondary processor. The assignment of a unit of work should proceed according to the algorithm detailed in Sect. 2.8
If the duty of this secondary processor was previously assigned, remove the highest priority subproblem from the priority queue of αBB problems and send it to this secondary processor.
Increment the number of αBB iterations
-
If the secondary processor has completed CSA work:
Receive the minimized conformation identified during the CSA search.
If this new conformation satisfies the acceptance criteria outlined in 2.8, add this new conformation to the CSA bank
If the number of CSA iterations since the last addition of conformers to the CSA bank exceeds a threshold value, add more conformers to the CSA bank from the queue of αBB solutions
Send out the next unit of CSA work to the current secondary processor. The assignment of a unit of work should proceed according to the algorithm detailed in Sect. 2.8
Increment the number of CSA iterations
-
If no results were waiting to be received by the primary processor:
If a conformer is not already undergoing perturbations with the idle cycles of the primary processor, randomly extract a conformation from the CSA bank
-
Perform a unit of work with the idle processor. A unit of work is defined by 10 repetitions of the following procedure:
Perturb the current conformation. The perturbation may be either a random perturbation of a single φ or ψ angle or a random shear movement of between 1 and 5 continuous residues. The shear movement is performed by perturbing the φ angle of a residue in one direction and perturbing the ψ angle of the previous residue with an equal magnitude but in the opposite direction. Both perturbations values are selected from a Gaussian distribution with a standard deviation of 0.5°
Evaluate the energy of the perturbed conformation
If the energy of the perturbed conformation is less than the original conformation, store the new conformation
If 500 units of work have been completed for a conformer and this new conformation satisfies the acceptance criteria outlined in Sect. 2.8, add this new conformation to the CSA bank
Send termination signals to all of the secondary nodes and receive the final results from each processor
3 Computational studies
The utility of an algorithm can be gauged by knowing both its strengths and its limitations. Previous algorithms have used global optimization-based approaches to identify low energy protein conformations using the ECEPP/3 force field [37, 40]. The proposed tertiary structure prediction algorithm will be evaluated using four protein test cases that have been studied in detail including published energy values, secondary structure predictions, distance and dihedral angle restraints, and root mean square deviations (RMSDs) of the Cα atoms from the native state. The computational studies described below were executed in parallel using 50 Intel 3.0 GHz Pentium processors for 72 hours on an available Beowulf cluster.
3.1 Immunoglobulin binding domain of protein G
The immunoglobulin binding domain of protein G from the Streptococcus species contains 56 amino acids and has shown unique properties, such as an extreme thermal stability. The structure of this protein has been determined using NMR spectroscopy [103] and X-ray crystallography [104]. This protein folds into a ββαββ motif, where the four β-strands form a single β-sheet and the single α-helix is located above this plane. The tightly-packed hydrophobic core and the extensive hydrogen bonding network make this protein an interesting and widely-used system for computational and theoretical studies.
The ASTRO-FOLD protein structure prediction approach was applied to protein G to further validate this ab initio approach [40]. The ASTRO-FOLD approach predicts a single α-helix between residues 23–34, so the φ and ψ dihedral angles of these residues are restricted to the range of [−85, −55] and [−50, −10], respectively. Klepeis and Floudas [40] correctly predicted the existence of four β-strands, located from residues 1–7, 16–21, 43–45, and 51–55. The φ and ψ dihedral angles of these β-strands are bounded from [−155, −75] and [110, 180], respectively. The hydrogen bonding network within the helix was enforced with 8 lower and upper distance bound restraints on Cα–Cα (i, i + 4) distances. The prediction of the β-sheet topology identified 12 β-sheet contacts that were enforced with lower and upper distance bounds of 4.5–6.5 Å on the Cα–Cα distances. Klepeis and Floudas [40] obtained φ and ψ dihedral angle bounds for the loop regions by additional free energy runs for oligopeptide segments. Their combined global optimization and torsion angle dynamics algorithm with these restraints yielded a protein conformation with an ECEPP/3 potential energy value of −267.0 kcal/mol and a Cα RMSD to the experimentally determined structure of 4.2 Å.
The restraints on the dihedral angles and distances described by Klepeis and Floudas [40] were approximately recreated for the purposes of comparing tertiary structure prediction algorithms for protein G. The specifics of the free energy runs for the oligopeptides representing the loop residues were not provided, so the comparison run was performed with unrestrained loop residues (i.e., φ and ψ bounds of [−180, 180]). Figure 1 summarizes the evolution of the results of the proposed hybrid tertiary structure prediction algorithm by plotting the minimum ECEPP/3 energy conformation achieved versus the number of CSA iterations that have been completed. The lowest energy conformation, with an ECEPP/3 potential energy of −418.254 kcal/mol, was identified after 2361 iterations of the CSA portion of the proposed hybrid global optimization algorithm.
Fig. 1.

The progression of the minimum ECEPP/3 potential energy conformation of protein G identified as a function of the number of iterations of the proposed tertiary structure prediction algorithm. Also marked for reference is the lowest energy previously achieved [40]
The quality of the ensemble of predicted protein conformers can be further evaluated by calculating their RMSDs from the native protein G structure. This evaluation of the ensemble is presented in Fig. 2. The conformer with the lowest energy in the ensemble has an RMSD of 4.97 Å from the native structure. The ensemble does contain several structures with a lower RMSD, including a conformer with an energy of −328.38 kcal/mol and a 2.81 Å RMSD from the native structure. Despite the significant population of low energy structures that can be found using the proposed hybrid tertiary structure prediction algorithm, the tertiary structure prediction method of Klepeis and Floudas [40] is unable to find structures within 100–200 kcal/mol of the minimum energy.
Fig. 2.

The ECEPP/3 potential energy of each protein conformer and its RMSD from the native protein G structure (PDB:2GB1). Also included for reference is the value reported in the literature [40]
The lowest energy conformer from the application of the proposed tertiary structure prediction algorithm is aligned to the native protein G structure in Fig. 3. There are differences in the exact placement of the secondary structure elements, especially the orientation of the α-helix with respect to the β-sheet, but the overall native topology is well-maintained in this lowest energy conformation.
Fig. 3.
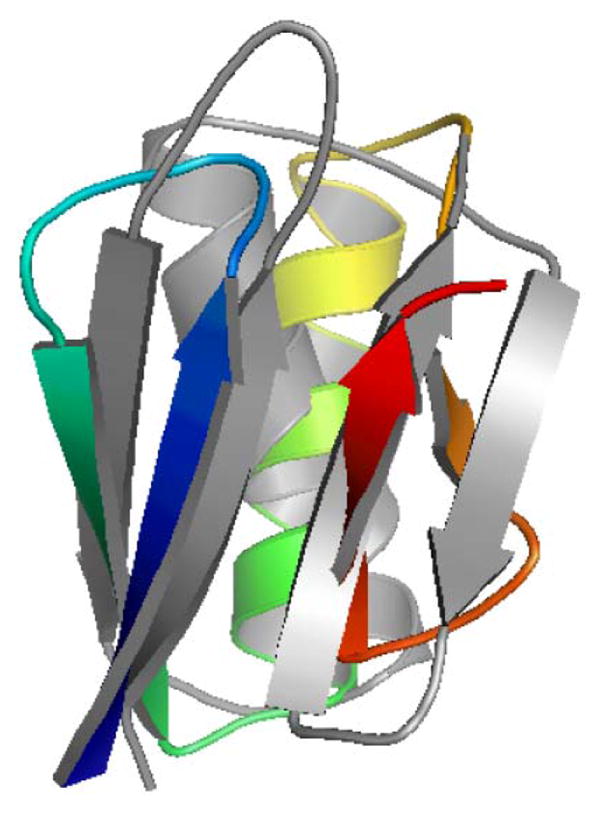
(Color online) An alignment of the lowest energy predicted conformation of protein G (color) to the native protein G structure (gray)
3.2 Target 59 from the CASP3 experiment
The target T59 was one of 43 protein sequences with unknown structure released during the CASP3 experiment. This sequence, representing the human Sm D3 protein, contains 75 amino acids. After the CASP experiment, this protein was later determined with a resolution of 2.0 Å using X-ray crystallography techniques [105]. The topology of this protein is similar to the common SH3 fold, which can be recognized by the antiparallel β-sheets that fold together to produce a barrel-like structure.
Klepeis and Floudas used the ASTRO-FOLD methodology to predict the structure of this protein [37]. The predicted location of the α-helical region for this protein is residues 6–11, which favorably overlaps the assignment of residues 6–13 as α-helical when using the experimental coordinates. The φ and ψ dihedral angles for these residues are restricted to the range of [−90, −40] and [−60, −10], respectively. This protein is predicted to have eight β-strands, spanning the residues 16–21, 26–29, 31–34, 39–43, 46–51, 53–57, 61–64, and 68–73. The φ and ψ dihedral angles of these residues with extended conformations are bounded by the ranges of [−180, −80] and [80, 180], respectively. The remaining residues, which define the loop regions between these secondary structure elements, are allowed to assume the entire range of possible φ and ψ angles, [−180, 180]. Klepeis and Floudas identified 30 lower and upper distance bounds to introduce on Cα–Cα distances representing the predicted β-sheet contacts. The combined global optimization and torsion angle dynamics algorithm of Klepeis and Floudas with these restraints yielded a protein conformation with an ECEPP/3 potential energy value of −395 kcal/mol and a Cα RMSD to the experimentally determined structure of 5.4 Å.
T59 represents yet another protein system to validate the application of the proposed tertiary structure prediction algorithm. Figure 4 describes the progress of this proposed hybrid algorithm by plotting the minimum ECEPP/3 energy conformation that has been identified versus the number of CSA iterations. The conformer with lowest ECEPP/3 energy, at a value of −582.858 kcal/mol, was identified by the algorithm after 480 iterations. The algorithm shows consistent progress for the first 483 iterations, indicating that further exploration of the conformational space may lead to structures with lower potential energies than the minimum value reported here. This observation was indeed validated by the identification of near-native protein structures with energies as low as −630 kcal/mol.
Fig. 4.

The progression of the minimum ECEPP/3 potential energy conformation of T59 identified as a function of the number of iterations of the proposed tertiary structure prediction algorithm. Also marked for reference is the lowest energy previously achieved [37]
The RMSD values of the predicted T59 conformers, identified by the proposed tertiary structure prediction algorithm, versus the T59 native structure are plotted as a function of their ECEPP/3 potential energy in Fig. 5. The conformer with the lowest energy in the ensemble has an ECEPP/3 potential energy of −582.858 kcal/mol and an RMSD to the native structure of 4.73 Å. The ensemble does contain several structures with a lower RMSD, including the conformer with the lowest RMSD of 3.43 Å and an energy of −545.47 kcal/mol. More than 3600 conformers are identified with an energy below −400 kcal/mol.
Fig. 5.

The ECEPP/3 potential energy of each protein conformer and its RMSD from the native T59 protein structure (PDB:1D3B). Also included for reference is the value reported in the literature [37]
Figure 6 illustrates the alignment of the lowest energy conformer from the application of the proposed tertiary structure algorithm versus the native T59 structure. There are differences in the exact placement of the secondary structure elements, but the overall native topology of this lowest energy conformation is well-maintained. The reasonable RMSD of 4.5 Å indicates that the differences easily identified by visual inspection do not contribute heavily to the structural deviations. Additional dihedral angle restraints on the loops or more extensive conformational sampling may lead to structures with lower energies and lower RMSDs from the native T59 structure.
Fig. 6.
(Color online) An alignment of the lowest energy predicted conformation of T59 (color) to the native T59 structure (gray)
3.3 Bovine pancreatic trypsin inhibitor (BPTI)
Serine protease inhibitors are responsible for regulating serine proteases like trypsin and chymotrypsin. The serine proteases are necessary for hydrolyzing peptides, which is an integral role for cell function. The bovine pancreatic trypsin inhibitor (BPTI) is a well-studied protein within the class of serine protease inhibitors. Its three-dimensional structure is characterized by 3 disulfide bonds (between Cysteine residues at positions 5–55, 14–38, and 30–51) that stabilize the protein in its native state. These disulfide bonds are conserved across the class of serine protease inhibitors. The tertiary structure of BPTI has been experimentally determined using standard X-ray crystallography techniques (PDB: 4PTI) [106] and by methods that combine X-ray and neutron diffraction experiments (PDB: 5PTI) [107].
BPTI was studied in detail by Klepeis and Floudas using the ASTRO-FOLD methodology [40]. The α-helical regions of the protein were determined to be between residues 2–5 and 47–54, whereas the residues 17–23, 29–35, and 44–46 were identified as β-strands. Klepeis and Floudas introduced φ and ψ dihedral angle bounds of [−85, −55] and [−50, −10] for the α-helical regions, bounds of [−155, −75] and [110, 180] for the β-strand regions, and unrestricted bounds ([−180, 180] and [−180, 180]) for the loop regions. For α-helical residues separated by four positions (i.e. positions i, i + 4) in the primary amino acid sequence, Cα–Cα distance bounds of 5.5–6.5 Å were introduced to enforce the formation of the intrahelical hydrogen bonding network. The topology of the β-sheets and disulfide bridges within BPTI were predicted using integer linear programming techniques [39] and enforced through additional distance restraints. For residues predicted to be contacts between two β-strands, Cα–Cα distance bounds of 4.5–6.5 Å were introduced. Finally, sulfur atoms of Cystine residues predicted to be in a disulfide bridge network are bounded by distances of 2.01–2.03 Å. The combined global optimization and torsion angle dynamics algorithm of Klepeis and Floudas with these restraints yielded a protein conformation with an ECEPP/3 potential energy value of −428.0 kcal/mol and a Cα RMSD to the experimentally determined structure of 4.1 Å.
The restraints described by [40] were recreated for the purposes of comparing tertiary structure prediction algorithms with a consistent basis. Figure 7 shows the energy values of the predicted protein conformers plotted against the RMSD values of these structure from the native structure of BPTI. Although the conformer with the lowest energy in the ensemble has an RMSD of 8.96 Å from the native structure, the ensemble contains several structures with a lower RMSD, including a conformer with an energy of −417.852 kcal/mol and a 3.41 Å RMSD from the native structure.
Fig. 7.

The ECEPP/3 potential energy of each protein conformer and its RMSD from the native BPTI structure (PDB:5PTI). Also included for reference are the values reported in the literature [40]
Figure 8 illustrates the alignment of the lowest energy conformer from the application of the proposed tertiary structure algorithm versus the native BPTI structure. The lowest energy conformation has the correct β-sheet topology, the correct disulfide bridge contacts, and the correct α-helical topology. However, there are significant differences in the placement of the α-helices relative to the β-strands.
Fig. 8.

(Color online) An alignment of the lowest energy conformation of BPTI (color) to the native BPTI structure (gray)
3.4 Target 114 from the CASP4 experiment
The target T114 was one of 43 protein sequences with unknown structure released during the CASP4 experiment. This 87 residue protein is an anti-fungal, chitin-binding protein from the organism Streptomyces tendae TÜ901. The structure of this protein was determined using multidimensional NMR spectroscopy techniques and released after the CASP4 experiment [108]. The structure of this protein contains 9 β-strands that pack together into 2 antiparallel β-sheets (one with 4 strands, one with 5 strands) that form a parallel β-sandwich motif.
Klepeis and Floudas closely examined the structure prediction of T114 as an evaluation of the ASTRO-FOLD protocol [37]. They predicted 7 β-strands, defined by the residues 12–16, 22–26, 31–37, 39–42, 48–54, 61–70, and 77–86, that were enforced by introducing φ and ψ dihedral angle bounds of [−180, −80] and [80, 180]. In addition, 34 β-sheet contacts were predicted and imposed as Cα–Cα distance bounds of 4.5–6.5 Å. A disulfide bridge was also predicted between residues 7 and 25, which bounded the distance between the sulfur atoms of these residues to a range of 2.01–2.03 Å. After applying their tertiary structure prediction algorithm, Klepeis and Floudas presented a structure with an energy of −530 kcal/mol and an RMSD to the native structure of 4.5 Å.
Figure 9 evaluates the RMSD to the native T114 structure for each of the conformers identified by the proposed tertiary structure prediction algorithm as a function of their ECEPP/3 potential energy. The lowest energy structure, with an RMSD to the native structure of 6.24 Å, has an energy value of −744.31 kcal/mol. A number of conformers with a better RMSD value exist in the ensemble, including a conformer with an energy value of −736.43 and an RMSD value of 4.18 Å and also the conformer with the lowest RMSD value of 4.00 Å at an energy value of −534.66 kcal/mol. This figure also illustrates the location of the original prediction of Klepeis and Floudas. In total, the proposed tertiary structure prediction algorithm identifies 1750 conformers with an energy value better than −530 kcal/mol.
Fig. 9.

The ECEPP/3 potential energy of each protein conformer and its RMSD from the native T114 protein structure (PDB:1GH5). Also included for reference are the values reported in the literature [37]
The lowest energy conformer is aligned to the native T114 structure in Fig. 10. Although the prediction of the overall topology, guided by the predicted β-sheet contacts, is correct, there are noticeable differences between the native structure and the lowest energy conformer in the predicted ensemble in the relative locations of several of the β-strand elements.
Fig. 10.

(Color online) An alignment of the lowest energy predicted conformation of T114 (color) to the native T114 structure (gray)
4 Conclusions
A new hybrid global optimization algorithm for protein tertiary structure prediction and its parallel implementation were presented. This hybrid global optimization algorithm successfully combines the αBB deterministic optimization algorithm and the conformational space annealing approach with an effective local minimization strategy. The local minimization requires torsion angle dynamics to identify initial conformations, rotamer optimization to achieve rough but efficient energy gains, and sequential quadratic programming to further minimize a protein conformation. The proposed method was able to outperform a related algorithmic approach in the literature for a computational study of four available test proteins, leading to lower energy protein structures and larger conformations of low-energy structures.
Supplementary Material
Acknowledgments
CAF gratefully acknowledges financial support from the National Science Foundation (R01 GM52032), the National Institutes of Health (R24 GM069736), and the US EPA (GAD R 832721-010). This work has not been reviewed by and does not represent the opinions of the funding agencies.
Footnotes
Electronic supplementary material The online version of this article (http://dx.doi.org/10.1007/s10589-009-9277-y) contains supplementary material, which is available to authorized users.
References
- 1.Levinthal C. How to fold graciously. In: Debrunner P, Tsibris JCM, Münck E, editors. Mossbauer Spectroscopy in Biological Systems. University of Illinois Press; Urbana: 1969. pp. 22–24. [Google Scholar]
- 2.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181(4096):223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 3.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 4.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karplus K, Barret Ch, Hughey R. Hidden Markov models for detecting remote protein homologies. Bioinformatics. 1998;14(10):846–856. doi: 10.1093/bioinformatics/14.10.846. [DOI] [PubMed] [Google Scholar]
- 6.Rychlewski L, Jaroszewski L, Li W, Godzik A. Comparison of sequence profiles. strategies for structural predictions using sequence information. Proteome Sci. 2000;9:232–241. doi: 10.1110/ps.9.2.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Narayana SV, Argos P. Residue contacts in protein structures and implications for protein folding. Int J Pept Protein Res. 1984;24:25–39. doi: 10.1111/j.1399-3011.1984.tb00924.x. [DOI] [PubMed] [Google Scholar]
- 8.Bowie JU, Lüthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 9.Godzik A, Kolinski A, Skolnick J. Topology fingerprint approach to the inverse folding problem. J Mol Biol. 1992;227:227–238. doi: 10.1016/0022-2836(92)90693-e. [DOI] [PubMed] [Google Scholar]
- 10.Chothia C. One thousand families for the molecular biologist. Nature. 1992;357:543–544. doi: 10.1038/357543a0. [DOI] [PubMed] [Google Scholar]
- 11.Grant A, Lee D, Orengo C. Progress towards mapping the universe of protein folds. Genome Biol. 2004;5:107. doi: 10.1186/gb-2004-5-5-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones DT. GenTHREADER: An efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol. 1999;287:797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- 13.Skolnick J, Zhang Y, Arakaki AK, Kolinski A, Boniecki M, Szilágyi A, Kihara D. TOUCHSTONE: A unified approach to protein structure prediction. Proteins Struct Funct Bioinf. 2003;53:469–479. doi: 10.1002/prot.10551. [DOI] [PubMed] [Google Scholar]
- 14.Skolnick J, Kihara D, Zhang Y. Development and large scale benchmark testing of the PROSPECTOR_3 threading algorithm. Proteins Struct Funct Bioinf. 2004;56:502–518. doi: 10.1002/prot.20106. [DOI] [PubMed] [Google Scholar]
- 15.Xu Y, Xu D. Protein threading using PROSPECT: Design and evolution. Proteins Struct Funct Bioinf. 2000;40:343–354. [PubMed] [Google Scholar]
- 16.Xu J, Li M, Kim D, Xu Y. RAPTOR: Optimal protein threading by linear programming. J Bioinf Comput Biol. 2003;1:95–117. doi: 10.1142/s0219720003000186. [DOI] [PubMed] [Google Scholar]
- 17.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simons KT, Kooperberg C, Huang C, Baker D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J Mol Biol. 1997;268:209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- 19.Rohl CA, Strauss CEM, Chivian D, Baker D. Modeling structurally variable regions in homologous proteins with Rosetta. Proteins Struct Funct Bioinf. 2004;55:656–677. doi: 10.1002/prot.10629. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Skolnick J. Tertiary structure predictions on a comprehensive benchmark of medium to large size proteins. Biophys J. 2004;87:2647–2655. doi: 10.1529/biophysj.104.045385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Skolnick J. Automated structure prediction of weakly homologous proteins on a genomic scale. Proc Natl Acad Sci. 2004;101:7594–7599. doi: 10.1073/pnas.0305695101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Skolnick J. SPICKER: A clustering approach to identify near-native protein folds. J Comput Chem. 2004;25:865–871. doi: 10.1002/jcc.20011. [DOI] [PubMed] [Google Scholar]
- 23.Wu S, Skolnick J, Zhang Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 2007;5:17–26. doi: 10.1186/1741-7007-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia Y, Huang ES, Levitt M, Samudrala R. Ab initio construction of protein tertiary structure using a hierarchical approach. J Mol Biol. 2000;300:171–185. doi: 10.1006/jmbi.2000.3835. [DOI] [PubMed] [Google Scholar]
- 25.Kussel E, Shimada J, Shakhnovich EI. A structure-based method for derivation of all-atom potentials for protein folding. Proc Natl Acad Sci. 2002;999:5343–5348. doi: 10.1073/pnas.072665799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozkan SB, Wu GA, Chodera JD, Dill KA. Protein folding by zipping and assembly. Proc Natl Acad Sci. 2007;104:11987–11992. doi: 10.1073/pnas.0703700104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srinivasan R, Rose GD. LINUS: A hierarchic procedure to predict the fold of a protein. Proteins Struct Funct Gen. 1995;22:81–89. doi: 10.1002/prot.340220202. [DOI] [PubMed] [Google Scholar]
- 28.Srinivasan R, Rose GD. Ab initio prediction of protein structure using LINUS. Proteins Struct Funct Bioinf. 2002;47:489–495. doi: 10.1002/prot.10103. [DOI] [PubMed] [Google Scholar]
- 29.Zagrovic B, Snow CD, Shirts MR, Pande VS. Simulation of folding of a small alpha-helical protein in atomistic detail using worldwide-distributed computing. J Mol Biol. 2002;323:927–937. doi: 10.1016/s0022-2836(02)00997-x. [DOI] [PubMed] [Google Scholar]
- 30.Liwo A, Arlukowicz P, Czaplewski C, Oldziej S, Pillardy J, Scheraga HA. A method for optimizing potential-energy functions by hierarchical design of the potential-energy landscape: Application to the UNRES force field. Proc Natl Acad Sci. 2002;99:1937–1942. doi: 10.1073/pnas.032675399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liwo A, Oldziez S, Pincus MR, Wawak RJ, Rackovsky S, Scheraga HA. A united-residue force field for off-lattice protein-structure simulations. I Functional forms and parameters of long-range side-chain interaction potentials from protein crystal data. J Comput Chem. 1997;18:849–873. [Google Scholar]
- 32.Liwo A, Pincus MR, Wawak RJ, Rackovsky S, Oldziej S, Scheraga HA. A united-residue force field for off-lattice protein structure simulations. II Parameterization of short-range interactions and determination of weights of energy terms by z-score optimization. J Comput Chem. 1997;18:874–887. [Google Scholar]
- 33.Lee J, Scheraga HA, Rackovsky S. New optimization method for conformational energy calculations on polypeptides: Conformational space annealing. J Comput Chem. 1997;18:1222–1232. [Google Scholar]
- 34.Lee J, Pillardy J, Czaplewski C, Arnautova Y, Ripoll DR, Liwo A, Gibson KD, Wawak RJ, Scheraga HA. Efficient parallel algorithms in global optimization of potential energy functions for peptides, proteins and crystals. Comput Phys Commun. 2000;128:399–411. [Google Scholar]
- 35.Czaplewski C, Liwo A, Pillardy J, Oldziej S, Scheraga HA. Improved conformational space annealing method to treat beta-structure with the UNRES force-field and to enhance scalability of parallel implementation. Polymer. 2004;45:677–686. [Google Scholar]
- 36.Nanias M, Chinchio M, Oldziej S, Czaplewski C, Scheraga HA. Protein structure prediction with the UNRES force-field using replica-exchange Monte Carlo-with-minimization; comparison with MCM, CSA, and CFMC. J Comput Chem. 2005;26:1472–1486. doi: 10.1002/jcc.20286. [DOI] [PubMed] [Google Scholar]
- 37.Klepeis JL, Floudas CA. ASTRO-FOLD: A combinatorial and global optimization framework for ab initio prediction of three-dimensional structures of proteins from the amino acid sequence. Biophys J. 2003;85:2119–2146. doi: 10.1016/s0006-3495(03)74640-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klepeis JL, Floudas CA. Ab initio prediction of helical segments in polypeptides. J Comput Chem. 2002;23(2):245–266. doi: 10.1002/jcc.10002. [DOI] [PubMed] [Google Scholar]
- 39.Klepeis JL, Floudas CA. Prediction of beta-sheet topology and disulfide bridges in polypeptides. J Comput Chem. 2003;24:191–208. doi: 10.1002/jcc.10167. [DOI] [PubMed] [Google Scholar]
- 40.Klepeis JL, Floudas CA. Ab initio tertiary structure prediction of proteins. J Glob Optim. 2003;25:113–140. [Google Scholar]
- 41.Klepeis JL, Pieja MT, Floudas CA. A new class of hybrid global optimization algorithms for peptide structure prediction: Integrated hybrids. Comput Phys Commun. 2003;151:121–140. [Google Scholar]
- 42.Klepeis JL, Pieja MT, Floudas CA. Hybrid global optimization algorithms for protein structure prediction: Alternating hybrids. Biophys J. 2003;84:869–882. doi: 10.1016/S0006-3495(03)74905-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klepeis JL, Wei YN, Hecht MH, Floudas CA. Ab initio prediction of the three-dimensional structure of a de novo designed protein: A double-blind case study. Proteins Struct Funct Bioinf. 2005;58:560–570. doi: 10.1002/prot.20338. [DOI] [PubMed] [Google Scholar]
- 44.Dunbrack RL. Sequence comparison and protein structure prediction. Curr Opin Struct Biol. 2006;16:374–384. doi: 10.1016/j.sbi.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 45.Bujnicki JM. Protein structure prediction by recombination of fragments. Chem Bio Chem. 2006;7:19–27. doi: 10.1002/cbic.200500235. [DOI] [PubMed] [Google Scholar]
- 46.Floudas CA, Fung HK, McAllister SR, Mönnigmann M, Rajgaria R. Advances in protein structure prediction and de novo protein design: A review. Chem Eng Sci. 2006;61:966–988. [Google Scholar]
- 47.Floudas CA. Computational methods in protein structure prediction. Biotechnol Bioeng. 2007;97:207–213. doi: 10.1002/bit.21411. [DOI] [PubMed] [Google Scholar]
- 48.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 49.MacKerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, III, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 50.Momany FA, McGuire RF, Burgess AW, Scheraga HA. Energy parameters in polypeptides. VII Geometric parameters, partial atomic charges, nonbonded interactions, hydrogen bond interactions, and intrinsic torsional potentials for the naturally occurring amino acids. J Phys Chem. 1975;79:2361–2381. [Google Scholar]
- 51.Némethy G, Gibson KD, Palmer KA, Yoon CN, Paterlini G, Zagari A, Rumsey S, Scheraga HA. Energy parameters in polypeptides. 10 Improved geometrical parameters and nonbonded interactions for use in the ECEPP/3 algorithm, with application to proline-containing peptides. J Phys Chem. 1992;96:6472–6484. [Google Scholar]
- 52.Arnautova YA, Jagielska A, Scheraga HA. A new force field (ECEPP-05) for peptides, proteins, and organic molecules. J Phys Chem B. 2006;110:5025–5044. doi: 10.1021/jp054994x. [DOI] [PubMed] [Google Scholar]
- 53.Ortiz AR, Kolinski A, Skolnick J. Fold assembly of small proteins using Monte Carlo simulations driven by restraints derived from multiple sequence alignments. J Mol Biol. 1998;277(2):419–448. doi: 10.1006/jmbi.1997.1595. [DOI] [PubMed] [Google Scholar]
- 54.McAllister SR, Mickus BE, Klepeis JL, Floudas CA. A novel approach for alpha-helical topology prediction in globular proteins: Generation of interhelical restraints. Proteins Struct Funct Bioinf. 2006;65:930–952. doi: 10.1002/prot.21095. [DOI] [PubMed] [Google Scholar]
- 55.Klepeis JL, Floudas CA. Analysis and prediction of loop segments in protein structures. Comput Chem Eng. 2005;29:423–436. [Google Scholar]
- 56.Mönnigmann M, Floudas CA. Protein loop structure prediction with flexible stem geometries. Proteins Struct Funct Bioinf. 2005;61:748–762. doi: 10.1002/prot.20669. [DOI] [PubMed] [Google Scholar]
- 57.Creighton TE. Proteins: Structures and Molecular Properties. 2. Freeman; New York: 1993. [Google Scholar]
- 58.Bazaraa MS, Sherali HD, Shetty CM. Nonlinear Programming: Theory and Algorithms. 2. Wiley; New York: 1993. [Google Scholar]
- 59.Fletcher R. Practical Methods of Optimization. 2. Wiley; New York: 1987. [Google Scholar]
- 60.Gill PE, Murray W, Wright MH. Practical Optimization. Academic Press; Burlington: 1981. [Google Scholar]
- 61.Gill PE, Murray W, Saunders M, Wright MH. NPSOL 4.0 User’s Guide. Systems Optimization Laboratory, Department of Operations Research, Standford University, CA; 1986. [Google Scholar]
- 62.Blumenthal LM. Theory and Applications of Distance Geometry. Cambridge University Press; Cambridge: 1953. [Google Scholar]
- 63.Crippen GM. A novel approach to the calculation of conformation: Distance geometry. J Comput Phys. 1977;26:449–452. [Google Scholar]
- 64.Crippen GM, Havel TF. Distance Geometry and Molecular Conformation. Wiley; New York: 1988. [Google Scholar]
- 65.Moré JJ, Wu Z. Distance geometry optimization for protein structures. J Glob Optim. 1999;15:219–234. [Google Scholar]
- 66.Allen MP, Tildesley DJ. Computer Simulation of Liquids. Clarendon Press; Oxford: 1987. [Google Scholar]
- 67.Brünger AT. X-PLOR, Version 3.1. A System for X-Ray Crystallography and NMR. Yale University Press; New Haven: 1992. [Google Scholar]
- 68.Braun W, Go N. Calculation of protein conformations by proton-proton distance constraints. A new efficient algorithm. J Mol Biol. 1985;186:611–626. doi: 10.1016/0022-2836(85)90134-2. [DOI] [PubMed] [Google Scholar]
- 69.Güntert P, Wüthrich K. Improved efficiency of protein structure calculations from NMR data using the program DIANA with redundant dihedral angle constraints. J Biomol NMR. 1991;1:446–456. doi: 10.1007/BF02192866. [DOI] [PubMed] [Google Scholar]
- 70.Jain A, Vaidehi N, Rodriguez G. A fast recursive algorithm for molecular dynamics simulation. J Comput Phys. 1993;106:258–268. [Google Scholar]
- 71.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 72.Stein EG, Rice LM, Brünger AT. Torsion angle dynamics as a new efficient tool for NMR structure calculation. J Magn Reson. 1997;124:154–164. doi: 10.1006/jmre.1996.1027. [DOI] [PubMed] [Google Scholar]
- 73.Güntert P. Structure calculation of biological macromolecules from NMR data. Q Rev Biophys. 1998;31:145–237. doi: 10.1017/s0033583598003436. [DOI] [PubMed] [Google Scholar]
- 74.Ponder JW, Richard FM. Tertiary templates for proteins. use of packing criteria in the enumeration of allowed sequences for different structural classes. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 75.Dunbrack RL, Karplus M. Backbone-dependent rotamer library for proteins. Application to side-chain prediction. J Mol Biol. 1993;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- 76.Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins Struct Funct Gen. 2000;40:389–408. [PubMed] [Google Scholar]
- 77.Dunbrack RL. Rotamer libraries in the 21st century. Curr Opin Struct Biol. 2002;12:431–440. doi: 10.1016/s0959-440x(02)00344-5. [DOI] [PubMed] [Google Scholar]
- 78.Desmet J, De Maeyer M, Hazes B, Lasters I. The dead-end elimination theorem and its use in protein sidechain positioning. Nature. 1992;356:539–542. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- 79.Looger LL, Hellinga HW. Generalized dead-end elimination algorithms make large-scale protein side-chain prediction tractable: Implications for protein design and structural genomics. J Mol Biol. 2001;307:429–445. doi: 10.1006/jmbi.2000.4424. [DOI] [PubMed] [Google Scholar]
- 80.Leach AR, Lemon AP. Exploring the conformational space of protein side chains using dead-end elimination and the A* algorithm. Proteins Struct Funct Gen. 1998;33:227–239. doi: 10.1002/(sici)1097-0134(19981101)33:2<227::aid-prot7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 81.Xie W, Sahinidis NV. Residue-rotamer-reduction algorithm for the protein side-chain conformation problem. Bioinformatics. 2006;22:188–194. doi: 10.1093/bioinformatics/bti763. [DOI] [PubMed] [Google Scholar]
- 82.Eriksson O, Zhou Y, Elofsson A. Side chain-positioning as an integer programming problem. WABI ‘01: Proceedings of the First International Workshop on Algorithms in Bioinformatics. 2001:128–141. [Google Scholar]
- 83.Kingsford CL, Chazelle B, Singh M. Solving and analyzing side-chain positioning problems using linear and integer programming. Bioinformatics. 2005;21:1028–1036. doi: 10.1093/bioinformatics/bti144. [DOI] [PubMed] [Google Scholar]
- 84.Canutescu AA, Shelenkov AA, Dunbrack RL. A graph-theory algorithm for rapid protein side-chain prediction. Proteome Sci. 2003;12:2001–2014. doi: 10.1110/ps.03154503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Holm L, Sander C. Fast and simple Monte Carlo algorithm for side-chain optimization in proteins: Application to model building by homology. Proteins Struct Funct Gen. 1992;14:213–223. doi: 10.1002/prot.340140208. [DOI] [PubMed] [Google Scholar]
- 86.Liang S, Grishin NV. Side-chain modeling with an optimized scoring function. Proteome Sci. 2002;11:322–331. doi: 10.1110/ps.24902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xiang Z, Honig B. Extending the accuracy limits of prediction for side-chain conformations. J Mol Biol. 2001;311:421–430. doi: 10.1006/jmbi.2001.4865. [DOI] [PubMed] [Google Scholar]
- 88.Tufféry P, Etchebest C, Hazout S, Lavery R. A new approach to the rapid determination of protein side chain conformations. J Biomol Struct Dyn. 1991;8:1267–1289. doi: 10.1080/07391102.1991.10507882. [DOI] [PubMed] [Google Scholar]
- 89.Lee C. Predicting protein mutant energetics by self-consistent ensemble optimization. J Mol Biol. 1994;236:918–939. doi: 10.1006/jmbi.1994.1198. [DOI] [PubMed] [Google Scholar]
- 90.Desmet J, Spriet J, Lasters I. Fast and accurate side-chain topology and energy refinement as a new method for protein structure optimization. Proteins Struct Funct Gen. 2002;48:31–43. doi: 10.1002/prot.10131. [DOI] [PubMed] [Google Scholar]
- 91.Levitt M, Gerstein M, Huang E, Subbiah S, Tsai J. Protein folding: The endgame. Annu Rev Biochem. 1997;66:549–579. doi: 10.1146/annurev.biochem.66.1.549. [DOI] [PubMed] [Google Scholar]
- 92.Baker D. Prediction and design of macromolecular structures and interactions. Philos Trans R Soc B. 2006;361:459–463. doi: 10.1098/rstb.2005.1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Adjiman CS, Androulakis IP, Maranas CD, Floudas CA. A global optimization method, αBB, for process design. Comput Chem Eng. 1996;20:S419–S424. [Google Scholar]
- 94.Adjiman CS, Androulakis IP, Floudas CA. Global optimization of MINLP problems in process synthesis and design. Comput Chem Eng. 1997;21:S445–S450. [Google Scholar]
- 95.Adjiman CS, Dallwig S, Floudas CA, Neumaier A. A global optimization method for general twice-differentiable NLPs. i Theoretical advances. Comput Chem Eng. 1998;22:1137–1158. [Google Scholar]
- 96.Adjiman CS, Androulakis IP, Floudas CA. A global optimization method for general twice-differentiable NLPs. ii Implementation and computational results. Comput Chem Eng. 1998;22:1159–1179. [Google Scholar]
- 97.Androulakis IP, Maranas CD, Floudas CA. αBB: A global optimization method for general constrained nonconvex problems. J Glob Optim. 1995;7:337–363. [Google Scholar]
- 98.Floudas CA. Nonconvex Optimization and its Applications. Kluwer Academic; Dordrecht: 2000. Deterministic Global Optimization: Theory, Methods and Applications. [Google Scholar]
- 99.Lee J, Scheraga HA, Rackovsky S. Conformational analysis of the 20-residue membrane-bound portion of melittin by conformational space annealing. Biopolymers. 1998;46:103–115. doi: 10.1002/(SICI)1097-0282(199808)46:2<103::AID-BIP5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 100.Lee J, Scheraga HA. Conformational space annealing by parallel computations: Extensive conformational search of metenkephalin and the 20-residue membrane-bound portion of melittin. Int J Quant Chem. 1999;75:255–265. [Google Scholar]
- 101.Ripoll D, Liwo A, Scheraga HA. New developments of the electrostatically driven Monte Carlo method: Tests on the membrane-bound portion of melittin. Biopolymers. 1998;46:117–126. doi: 10.1002/(SICI)1097-0282(199808)46:2<117::AID-BIP6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 102.Kirkpatrick S, Gelatt CD, Jr, Vecchi MP. Optimization by simulated annealing. Science. 1983;220:671–680. doi: 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- 103.Gronenborn AM, Filpula DR, Essig NZ, Achari A, Whitlow M, Wingfield PT, Clore GM. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science. 1991;253:657–661. doi: 10.1126/science.1871600. [DOI] [PubMed] [Google Scholar]
- 104.Gallagher T, Alexander P, Bryan P, Gilliland GL. Two crystal structures of the B1 immunoglobulin-binding domain of streptococcal protein G and comparison with NMR. Biochem. 1994;33:4721–4729. [PubMed] [Google Scholar]
- 105.Kambach C, Walke S, Young R, Avis JM, de la Fortelle E, Raker VA, Lührmann R, Li J, Nagai K. Crystal structures of two Sm protein complexes and their implications for the assembly of the spliceosomal snRNPs. Cell. 1999;96:375–387. doi: 10.1016/s0092-8674(00)80550-4. [DOI] [PubMed] [Google Scholar]
- 106.Deisenhofer J, Steigemann W. Crystallographic refinement of structure of bovine pancreatic trypsin-inhibitor at 1.5 Å resolution. Acta Crystallogr Sect B. 1975;31:238–250. [Google Scholar]
- 107.Wlodawer A, Walter J, Huber R, Sjolin L. Structure of bovine pancreatic trypsin-inhibitor: Results of joint neutron and x-ray refinement of crystal form II. J Mol Biol. 1984;180:301–329. doi: 10.1016/s0022-2836(84)80006-6. [DOI] [PubMed] [Google Scholar]
- 108.Campos-Olivas R, Hörr I, Bormann C, Jung G, Gronenborn AM. Solution structure, backbone dynamics and chitin binding properties of the anti-fungal protein from Streptomyces tendae TÜ901. J Mol Biol. 2001;308:765–782. doi: 10.1006/jmbi.2001.4622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.