

Abstract
Despite the association of prevalent health conditions with coronavirus disease 2019 (COVID-19) severity, the disease-modifying biomolecules and their pathogenetic mechanisms remain unclear. This study aimed to understand the influences of COVID-19 on different comorbidities and vice versa through network-based gene expression analyses. Using the shared dysregulated genes, we identified key genetic determinants and signaling pathways that may involve in their shared pathogenesis. The COVID-19 showed significant upregulation of 93 genes and downregulation of 15 genes. Interestingly, it shares 28, 17, 6 and 7 genes with diabetes mellitus (DM), lung cancer (LC), myocardial infarction and hypertension, respectively. Importantly, COVID-19 shared three upregulated genes (i.e. MX2, IRF7 and ADAM8) with DM and LC. Conversely, downregulation of two genes (i.e. PPARGC1A and METTL7A) was found in COVID-19 and LC. Besides, most of the shared pathways were related to inflammatory responses. Furthermore, we identified six potential biomarkers and several important regulatory factors, e.g. transcription factors and microRNAs, while notable drug candidates included captopril, rilonacept and canakinumab. Moreover, prognostic analysis suggests concomitant COVID-19 may result in poor outcome of LC patients. This study provides the molecular basis and routes of the COVID-19 progression due to comorbidities. We believe these findings might be useful to further understand the intricate association of these diseases as well as for the therapeutic development.
Keywords: COVID-19 pandemic, SARS-CoV-2, RNA-Seq, hypertension, microarray, comorbidities
Introduction
The ongoing COVID-19 pandemic has plagued the entire world [1–4] with a 3.55% global case fatality rate (CFR), as of 17 August 2020 (Source: Statista). The etiological agent of this international outbreak is an RNA virus known as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that belongs to the Coronaviridae family [1, 2, 5]. Depending on the demography and environments, the spectrum of clinical manifestations ranges from the common cold to respiratory failure [6–9]. Importantly, the persistence and prognosis of COVID-19 are greatly influenced by the underlying health conditions and age of the patients [10, 11]. Two or more coexisting health conditions are comorbidities when they influence each other with their shared pathogenesis [12, 13]. Remarkably, the most frequent comorbidities in COVID-19 are hypertension (HT) (27–30%), diabetes (19%) and coronary heart disease (6–8%) [5, 8, 11, 14]. Their associations may lead to the discovery of other potential risk factors that might help in identifying and managing high-risk populations [11, 15]. Although comorbidities are associated with the severity of COVID-19, the specific disease-modifying mechanisms need to be investigated [8].
The clinical manifestations of COVID-19 occur once the Spike (S) protein of SARS-CoV-2 binds to its cell surface receptor angiotensin-converting enzyme 2 (ACE2) that activates the innate immune system, leading to catastrophic cytokine storms, excessive release of pro-inflammatory cytokines and chemokines [16–19]. ACE2 expresses in most metabolic tissues and organs, including the lung, pancreas, intestine and kidneys [19]. Recent studies revealed HT as one of the most frequent comorbidities in COVID-19 patients [5]. In a study, about 23.4% of the severe COVID-19 patients (N = 1099) were reported to have HT [20]. Previously, clinical studies suggested HT as a risk factor for high death rates in SARS and MERS as well [21]. Patients with HT and cardiovascular disease are usually treated with ACE inhibitors and angiotensin receptor blockers (ARBs) that increase the expression of ACE2 which in turn attracts more viral load [22]. Therefore, disease severity may arise due to the role of ACE2 in the pathogenesis of HT and cardiovascular diseases [5]. Though the coexistence of HT and SARS-CoV-2 showed a higher mortality rate, there is insufficient evidence to establish the link between the prevalence of HT and the susceptibility for SARS-CoV-2 infection [5]. Hence, the specific molecular mechanism of high blood pressure that may be responsible for the severity of COVID-19 is yet to be studied.
Being the most prevalent global disease, diabetes has become the most common risk factor in COVID-19 patients [23]. Nationwide analysis in China showed that 34.6% of diabetic patients have developed a severe form of COVID-19 [24]. However, current data indicate that diabetic individuals are not prone to be infected with SARS-CoV-2 compared to the general population; rather, it could provoke the severity of COVID-19 [19, 25, 26]. Although earlier studies were focused on type 2 diabetes, recent studies suggested that COVID-19 patients with type 1 diabetes could also be at a potential risk of disease severity [27]. As mentioned earlier, ACE2 receptors are expressed in metabolic organs/tissues/cells, for example, pancreatic beta cells associated with controlling glucose metabolism; hence, entry of SARS-CoV-2 may lead to altered glucose metabolism, which possibly complicates the pathophysiology of predominant diabetes [19, 27, 28]. Furthermore, patients with lung cancer (LC) are prone to the severity of COVID-19 [29, 30]. In a homogenous study, a significant spike in CFR (52.3%) has been observed in LC patients with COVID-19 infection as compared to the general population [31]. Considering the predisposition of lung infection and compromised immunity in LC patients, the higher CFR from COVID-19 in LC patients is not surprising. Therefore, identification of the genetic determinants that may have worsened the LC outcome is important for optimal care during this pandemic. Hence, genetic determinants that are triggered by the COVID-19 and lead to a poor prognosis should be determined.
In our previous study, we determined the pathogenetic profile and comorbidities related to COVID-19 and SARS-like viruses [32]. We identified different risk factors and biomarkers involved in the disease progression. Therefore, this study aims to reveal the molecular basis for the prevalence of COVID-19 with the four major comorbidities (i.e. diabetes, cardiovascular disease, HT and LC) as well as the routes of their shared pathogenesis following the workflow depicted in Figure 1. Functional enrichment of their shared pathogenesis will provide a specific disease-modifying mechanism that may link these comorbidities with COVID-19 progression and vice versa.
Figure 1.
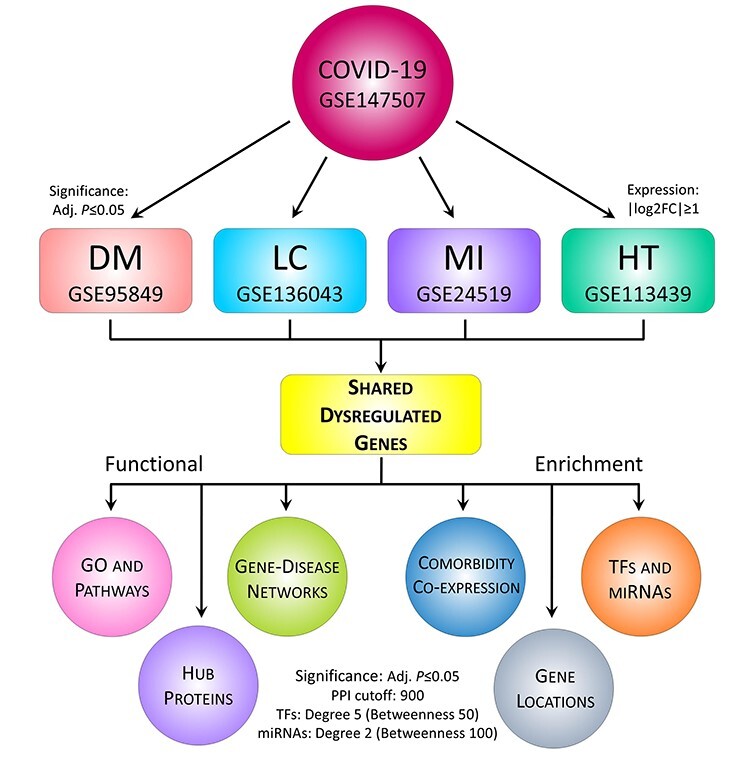
The proposed workflow for deciphering genetic determinants in COVID-19 pathophysiology. The RNA-Seq and microarray datasets of COVID-19 and four other diseases (i.e. DM, MI, LC and HT) were collected and analyzed for comparative gene expression analysis. In doing so, R package DSeq2 and Limma were applied for RNA-Seq normalization and differential expression, respectively. Herein, log2FC ≥ |1| was used to identify DEGs, and statistical significance was set to adjusted P-value ≤ 0.05. Next, the DEGs were subjected to functional enrichment in terms of distributions, GO and pathways, comorbidities, PPI, regulatory biomarkers, PDI and survival analysis. For PPI, the network was constructed with STRING confidence cutoff of 900 and potential hubs were identified using five different methods, while regulatory factors (e.g. TFs and miRNAs) were determined with degree centrality of 5 and 2, respectively. To detect the top-ranked factors, additional filtering was done using betweenness centrality of 50 and 100, respectively. For pathways, we considered Reactome (2016), KEGG (human; 2019) and WikiPathways (human; 2019) databases, while GO terms were determined with Biological Process (2018), Cellular Component (2018) and Molecular Function (2018) databases. In both cases, the adjusted P-value ≤ 0.05 was considered statistically significant.
Materials and methods
Datasets
To assess the impact of COVID-19 and its genetic association with other prevalent diseases, we retrieved and analyzed the relevant RNA-Seq and microarray datasets from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/). We utilized the gene expression data of COVID-19 and four relevant diseases i.e. diabetes mellitus (DM), LC, myocardial infarction (MI) and HT. In total, five different datasets with accession numbers GSE147507, GSE95849, GSE136043, GSE24519 and GSE113439 were used in this study [33–36]. The COVID-19 dataset (GSE147507) is an Illumina NextSeq 500 (Homo sapiens) platform-derived RNA-Seq data of lung epithelial cells treated with SARS-CoV-2 and healthy tissues in triplicates. The diabetes dataset (GSE95849) is a gene expression profile of six DM patients and six normal individuals, which has been developed using the Phalanx Human lncRNA OneArray v1_mRNA platform. The LC dataset (GSE136043) is a messenger ribonucleic acid (RNA) (mRNA) microarray using Agilent-026652 Whole Human Genome Microarray 4x44K v2, which includes five fresh tissues from LC patients and normal controls. The MI dataset (GSE24519) is an expression microarray that has been derived from 17 patients with their first acute MI compared with healthy individuals using the CodeLink™ Human Whole Genome Bioarray. Finally, the HT dataset (GSE113439) is a gene expression profile that has been derived from the lung tissue from 15 patients with pulmonary arterial HT and from 11 normal controls using the Affymetrix Human Gene v1.0 ST Array.
Analysis of differential expression and shared DEGs
In this study, we employed publicly available RNA-Seq and microarray datasets. Initially, the RNA-Seq data were normalized and converted to microarray equivalent using the DSeq2 R package to facilitate easy comparison. To identify differentially expressed genes (DEGs) in each dataset, we applied the Limma R package using two criteria, such as log2 fold change (FC) and adjusted P-value. We have considered log2FC ≥ 1 for upregulated genes and log2FC ≤ −1 for downregulated genes. Further, an adjusted P-value ≤ 0.05 was used to filter out significant DEGs. Next, to determine the shared DEGs, we compared COVID-19 dataset with four other selected diseases using Venny v2.1 web tool (https://bioinfogp.cnb.csic.es/tools/venny/). The gene–disease network (GDN) was created using bipartite graph theory [13, 32] and was visualized with Cytoscape v3.7 [37].
Distribution of DEGs and comorbidity profiling
Chromosomal location and expressional distribution of a gene are important to understand the pathophysiology of specific genes and to identify drug targets. Therefore, the chromosomal location of the DEGs was anticipated with the ShinyGO web tool [38]. Further, we utilized the PaGenBase dataset via the Metascape web server [39] for the tissue-specific distribution of DEGs. To determine the comorbidities associated with the DEGs, we used the DisGenNet database via the Metascape server [39]. Statistical significance was set to adjusted P-value ≤ 0.05. The expression pattern of the shared DEGs in other diseases was assessed with the Expression Atlas database [40]. The co-expression data were retrieved based on the log2FC of each gene for which disease versus normal datasets were considered. A clustered heatmap was created with the disease-wise expression value (log2FC) of shared DEGs using the Morpheus web tool (https://software.broadinstitute.org/morpheus/).
Enrichment of GO and signaling pathways
We predicted the signaling pathways and gene ontologies associated with the shared DEGs by using different databases via the Enrichr web server [41]. For pathways, we considered Reactome (2016), KEGG pathways (2019) and WikiPathways (2019) databases, while we considered biological process (2018), cellular component (2018) and molecular function (2018) for gene ontologies. The significant pathways were filtered with the adjusted P-value and the cutoff score was set to 0.05. The enrichment plots were visualized using the ImageGP web tool (http://www.ehbio.com/ImageGP/).
Analysis of protein–protein interaction
Protein–protein interaction (PPI) of the shared DEGs was analyzed using the STRING database via NetworkAnalyst v3.0 web server [42]. The PPI network was constructed by using the generic PPI option, where the organism was specified to H. sapiens, STRING with experimental evidence as the dataset and confidence score cutoff was set to 900. To determine potential hubs within the PPI network, we then applied different local- and global-based methods using cytoHubba plugin [43] in Cytoscape v3.7 [37]. While the local method ranked hubs based on the relationship between node and its direct neighbor, the global method ranked hubs based on the interaction between the node and the whole network [43]. In total, five different methods were considered, including three local rank methods, i.e. degree, maximum neighborhood component (MNC), maximal clique centrality (MCC), and two global rank methods, i.e. edge percolated component (EPC) and betweenness [43]. Next, we compared the results and identified the common nodes as the most potential hubs. Finally, the networks were customized with Cytoscape v3.7.
Identification of regulatory biomolecules
Regulatory molecules such as transcription factors (TFs) and microRNAs (miRNAs) are responsible for significant changes in transcription and expression outcomes. Therefore, we employed experimentally verified JASPAR [44] and miRTarbase v6.0 [45] datasets via the NetworkAnalyst v3.0 web tool [42] to anticipate TF–gene and miRNA–gene interactions. To eliminate non-major signature molecules, we filtered the TF–gene and miRNA–gene networks with degree centrality of 5 and 2, respectively. Both networks were customized with Cytoscape v3.7 [37].
Protein–drug interaction network
One of the primary objectives of this line of research focuses on pinpointing the potential drug molecules. With the shared DEGs, we determined the protein–drug interaction (PDI) network using NetworkAnalyst v3.0 web server [42] equipped with DrugBank v5.0. The network data were downloaded and customized with the Cytoscape v3.7 [37].
Survival analysis of the LC-associated genes
The effect of shared DEGs on LC patient’s survival was evaluated with PrognoScan server [46] and the survival plots were generated. The PrognoScan is a widely used server for survival analysis based on the genomics datasets from multiple cancers. It uses quickly confirmed disease prophecies, including overall survival (OS), relapse-free survival (RFS), distant metastasis-free survival (DMFS) and post-progression survival (PPS). The Cox P-value less than 0.05 was considered statistically significant.
Results
Differentially expression and distribution of DEGs
We analyzed and identified 108, 3022, 1532, 1361 and 618 significant DEGs (Adj. P ≤ 0.05) in COVID-19, DM, LC, MI and HT, respectively. Among them, the number of upregulated genes was 93, 2878, 722 and 853, respectively, while the number of downregulated genes was 15, 144, 810 and 508, respectively (Supplementary File S1-5). After comparing the COVID-19 with other datasets, we found 49 unique shared DEGs in which 42 genes were upregulated (Figure 2E) and only 7 genes (i.e. CXCL14, CYP4F3, MAP7D2, METTL7A, NANOS1, PPARGC1A and VTCN1) were downregulated (Supplementary File S6). To understand the pathogenetic involvement of COVID-19 with the aforesaid diseases, we performed a cross-comparative analysis among the gene expression profiles. The Venn diagram of Figure 2A shows that COVID-19 shares 28, 17, 6 and 7 genes with DM, LC, MI and HT. To visualize their association, we constructed a gene–disease relationship network (GDN) centered on the COVID-19 as shown in Figure 2E. Particularly, three genes (i.e. ADAM8, IRF7 and MX2) were upregulated among COVID-19, DM and LC, while a single gene SLC6A14 was common among COVID-19, MI and HT. Likewise, upregulation of TLR2 was observed in COVID-19, DM and HT. Further, CCL20 expression was shared with LC and HT. Conversely, COVID-19 shares downregulation of two genes (i.e. PPARGC1A and METTL7A) with LC and NANOS1 with MI.
Figure 2.

Comparison, distribution and comorbidities of shared dysregulated genes. Herein, (A) comparison of gene expression: (i) COVID-19 with DM and LC and (ii) COVID-19 with MI, and HT using Venn diagram, (B) chromosomal location of genes, (C) tissue- and cell-specific distribution of genes, (D) top-20 diseases associated with shared DEGs and (E) infectome–diseasome network where the octagon-shaped nodes represent five diseases, while circular nodes delineate the genes involved. Nodes with light violate color indicate upregulated genes and light blue indicate downregulated genes.
Targeting a protein at the transcription level requires the exact cellular and molecular locations of the respective genes as depicted in Figure 2B and C). Most of the shared DEGs (11) were located at chromosome 11, while chromosomes 1 and 12 contain 5 DEGs each. Rest are distributed throughout the genome except for 3, 9, 16, 18 and Y chromosomes. Shared DEGs were also absent in the mitochondrial (MT) genome (Figure 2B). The expressional distribution of DEGs throughout the cells and tissues reveals that most of the DEGs are expressed in lung tissue (12), followed by spleen (8), liver (6) and blood (5), while the lowest number of genes (3) is expressed in the bone marrow tissue and dorsal root ganglion (DRG) cells (Figure 2C; Supplementary File S7).
Expressions of DEGs in other diseases and comorbidities
Based on the gene–disease dataset, we identified the top 20 diseases that are highly relevant to our shared DEGs (Supplementary File S8). Figure 2D shows the number of genes involved with significant diseases. Herein, most of the shared DEGs were found to be related to causing liver cirrhosis (10), followed by influenza (8), colonic neoplasms (7) and other diseases. Importantly, five DEGs were found to be involved in rheumatoid arthritis, liver injury, DM, MI and reperfusion injury (Figure 2D). Out of a total of 49 shared DEGs, co-expression data of 48 genes were available in the Expression Atlas for 33 different health conditions as shown in Figure 3 (Supplementary File S9). Based on the heatmap, we found that the expression of shared DEGs varies with diseases. For instance, most DEGs were positively regulated in respiratory diseases, arthritis, psoriasis, glioblastoma, Crohn disease, ulcerative colitis, colorectal cancer, skin diseases, pneumonia, keratosis, lupus erythematosus, esophageal cancer, etc. as evidenced by the pink-colored cluster (Figure 3). Conversely, DEGs were negatively regulated in erythroleukemia, prostate, LC, pituitary disease, etc. as evidenced by the green clustered at the top of the heatmap (Figure 3).
Figure 3.
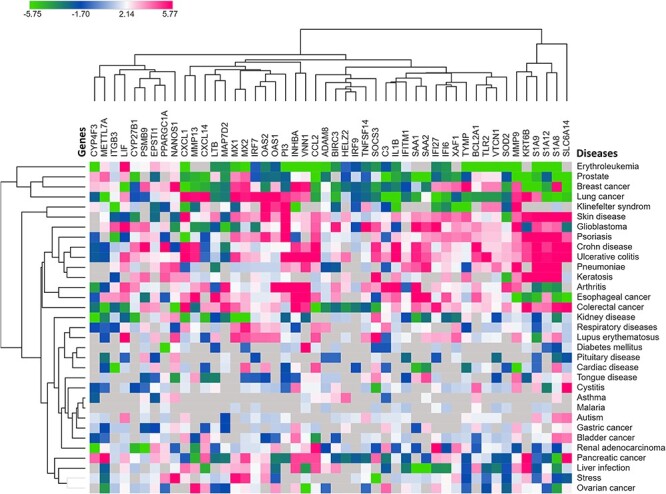
Heatmap showing the level of expression of shared DEGs in various diseases. Herein, green- and pink-colored boxes indicate the over- and underexpression of the genes in respective diseases, respectively, while the clustering feature shows the related co-expression of the genes based on the log2FC values of the shared DEGs.
Signaling pathways and gene ontologies
In a complex disease, a diverse range of signaling pathways and GO terms are involved in the orchestration and progression of diseases. In this process, we used 49 shared DEGs to determine significant pathways and gene ontologies that may link COVID-19 and the four considered diseases. Figures 4 and 5 show 30 significant pathways and 30 GO terms from three datasets (top-10 terms from each; Supplementary File S10-11). Pathways and GO terms were selected based on the number of genes involved and adjusted P-value less than or equal to 0.05. Most of the pathways are related to inflammatory responses (15) followed by bacterial/viral infections (9). Other pathways include angiogenesis/cancer (two), brain disease (two) and metabolisms (two) as shown in Figure 4. Top-10 pathways are immune system, cytokine signaling in the immune system, innate immune system, herpes simplex virus 1 infection, neutrophil degranulation, interferon-alpha/beta signaling, interferon signaling, signaling by interleukins, IL-17 signaling pathway and cytokine–cytokine receptor interaction (Figure 4).
Figure 4.

Signaling pathways involved with the shared dysregulated genes. For important pathways, we used Reactome (2016), KEGG (2019) and WikiPathways (2019) datasets and presented 30 most relevant pathways (top-10 from each of three databases) with adjusted P-value less than 0.05.
Figure 5.

Gene ontological groups related to the shared dysregulated genes. For GO terms, we used biological process (2018), cellular component (2018) and molecular function (2018) databases. The figure depicted the 30 most relevant GO terms (top-10 from each of three databases) with an adjusted P-value less than 0.05.
In addition to signaling pathways, over-presented GO groups were predicted for shared DEGs. Totally 30 GO terms were selected based on the number of genes and adjusted P-value less than or equal to 0.05. Top-10 GO terms were identified as immune system process (35), cellular response to chemical stimulus (34), immune response (33), response to stress (32), response to external stimulus (31), cell surface receptor signaling pathway (29), response to organic substance (29), defense response (28), regulation of signal transduction (28) and multi-organism process (27) as shown in Figure 5. These ontological features were common in SARS-CoV disease and COVID-19 complications. Therefore, they could either be risk factors or regulatory checkpoints in COVID-19 disease.
PPI network and hub-proteins
A PPI network has been built from the common DEGs’ interactions, which consists of 185 nodes and 201 edges (Figure 6A). We employed five methods to determine the hub-proteins, where each method identified the top-10 hub-nodes within the PPI network (Figure 6B–E). Interestingly, 6 out of 10 hub-proteins were common in all methods except for MNC (data not shown here). As anticipated by four different methods and exhibited a minimum of eight interconnections, we recognized six hub-nodes as potential hub-proteins, i.e. SOCS3, TLR2, BIRC3, PPARGC1A, HELZ2 and IRF7. The biological functions of these hub-proteins are tabulated in Table 1. Conversely, MNC method predicted only three proteins (i.e. MMP13, NFKB1 and LIF) from the shared DEGs as hubs that were not found by other methods.
Figure 6.

The PPI network. This network contains (A) a total of 185 proteins, including 13 shared DEGs in which hubs were predicted and indicated according to degree method using STRING database (confidence cutoff of 900). Four smaller networks are depicting hub-proteins anticipated by (B) degree, (C) MCC, (D) EPC and (E) betweenness methods. For all methods, top-10 hub-nodes are ranked with red- to yellow-colored gradient.
Table 1.
The name and biological significance of the potential hub-nodes
| Hub-Proteins | Name | Functions | UniProt ID | References |
|---|---|---|---|---|
| SOCS3 | Suppressors of cytokine signaling 3 | SOCS3 is a negative regulator of the JAK–STAT signaling pathway. It promotes macrophage polarization that plays a key role in lung inflammation as well as in LC | O14543 | [47] |
| TLR2 | Toll-like receptor 2 | TLR2 recognizes viral proteins and involves in APC activation. It may also recognize Spike of SARS-CoV-2. In type 1 diabetes, TLR2 signaling modulates CD4 + CD25+ Tregs and promotes inflammation to prevent diabetes | O60603 | [48, 49] |
| BIRC3 | Baculovial inhibitor of apoptosis (IAP) repeat-containing 3 | BIRC3 is a member of the IAP protein family. It plays a crucial role in NF-κB signaling pathway. It is upregulated in glioblastoma which causes therapeutic resistance | Q13489 | [50–52] |
| PPARGC1A | Peroxisome proliferator-activated receptor-gamma coactivator-1α | PPARGC1A plays a vital role in myocardial energy metabolism. In hypertensive individuals, the PPARGC1A Gly482Ser polymorphism is engaged in hypertrophy and diastolic dysfunction which may incline to heart failure. The upregulation of PPARGC1A also facilitates LC metastasis | Q9UBK2 | [53, 54] |
| HELZ2 | Helicase with zinc finger 2 | HELZ2 modulates the activity of IFN to counteract viral infection. Also, it is a transcription coactivator of PPAR-γ, and HELZ2-deficient mice exhibited improved insulin resistance through enhancing lipid burning in the liver | Q9BYK8 | [55, 56] |
| IRF7 | Interferon regulatory factor 7 | IRF7 is an authentic target of p53 to produce type 1 interferon upon viral infection. It is upregulated in COVID-19 and also facilitates the progression of LC | Q92985 | [57, 58] |
Transcriptional and post-transcriptional biomarkers
Using the shared DEGs, we found 45 miRNAs and 29 TFs that might influence the expression pattern of those genes and lead to the progression of diseases as depicted in Figure 7 and Supplementary File S12. Among all the miRNAs, we identified seven miRNAs (i.e. miR-98-5p, miR-146a-5p, miR-335-5p, miR-204–5p, miR-34a-5p, miR-26b-5p and miR-106b-5p) with betweenness centrality ≥ 100 (Figure 7A). These miRNAs may involve in the progression of COVID-19 and other diseases. Out of a total 29 TFs, the top 10 TFs (i.e. FOXC1, GATA2, NFKB1, YY1, PPARG, RELA, USF2, JUN, CREB1 and E2F1) were identified with betweenness centrality ≥ 50 as shown in Figure 7B. Apart from these, our study includes TFs and miRNAs that are highly relevant to COVID-19 and other disease progressions as described in the Discussion section.
Figure 7.

Gene regulatory networks associated with the shared dysregulated genes. The figure showing (A) gene–miRNA interacting network and (B) gene–TF interacting network. The interacting network of miRNAs and TFs were filtered with degree centrality less than and equal to 2 and 5, respectively. In these networks, hexagons are shared DEGs, while lime and blue circles represent associated miRNAs or TFs, respectively.
Potential drug candidates
Using the PDI approach, we found a total of 36 drug molecules acting against three proteins (i.e. IL1B, MMP9 and MMP13) out of 49 protein-coded genes. Figure 8 showed the interactions among the target proteins and drug candidates. Among all, 14 drugs showed antagonistic relationships to MMP13, followed by IL1B (13) and MMP9 (11). Only six drugs were found approved (i.e. rilonacept, canakinumab, gallium nitrate, minocycline, glucosamine and captopril), while others were either experimental (18) or investigational (12). Importantly, most of the molecules were anti-inflammatory drug candidates, such as rilonacept, canakinumab, gevokizumab, andrographolide, VX-702, etiprednol dicloacetate, SCIO-469 and dilmapimod.
Figure 8.
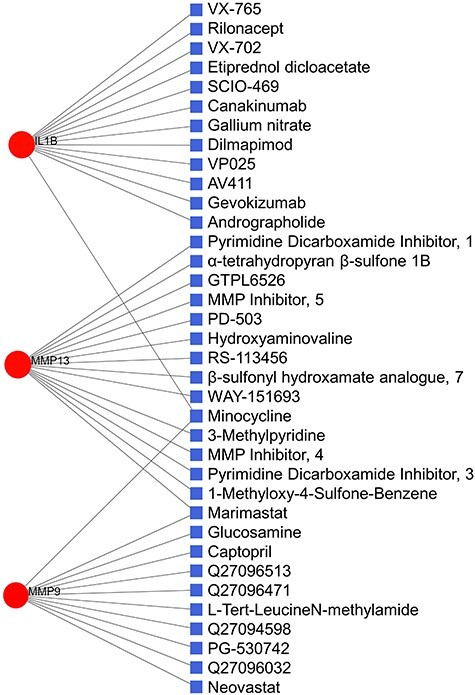
PDI network. Of the total 39 nodes, circles represent the shared dysregulated genes (red), while squares indicate the interacting drugs molecules (blue).
Effect of COVID-19 on the prognosis of LC patients
Out of 17 shared DEGs between COVID-19 and LC, significant patient survival statistics were found for 15 genes (Supplementary File S13). Figure 9 shows the probability of OS for each gene based on their positive and negative expressions. In most cases, the analysis revealed a significant positive correlation for the OS rate. For COVID-19-like expression, for instance, the chances of survival in LC patients tend to reduce due to the COVID-19-related expression of nine DEGs (i.e. ADAM8, C3, CCL20, CXCL14, FAM167A, METTL7A, PPARGC1A, SAA2 and TYMP), while the survival rate increases for five DEGs that include IFITM1, IRF7, LIF, MAP7D2 and VTCN1. The OS outcome was not affected by the MX2 gene regardless of its dysregulation. Therefore, the results suggested that genetic expression in COVID-19 may lead to a poor prognosis in LC patients.
Figure 9.

The relationship between the expression of shared DEGs and survival of LC patients. In survival curves, red- and blue-colored lines represent overexpression and underexpression of the respective genes in patients’ survival, where (A–O) show OS of shared genes between COVID-19 and LC. Herein, five genes, such as CXCL14, MAP7D2, METTL7A, PPARGC1A and VTCN1, are downregulated in COVID-19 and the rest are upregulated.
Discussion
In this study, we analyzed and compared gene expression data of COVID-19 and four related comorbidities to understand their shared pathogenesis leading to the severity of SARS-CoV-2 infection. We found that COVID-19 showed higher relevance to DM and LC as suggested by their shared gene expression. Besides, six key proteins were identified, which are central to COVID-19 and associated comorbidities. Furthermore, the shared DEGs determined in this study were found to have an impact on the survival of LC patients. Finally, we identified notable gene regulatory components, such as TFs and miRNAs, that essentially control the major pathways involved in these diseases.
Based on the comparative analysis, the COVID-19 transcriptome revealed 49 DEGs that are common to the considered four comorbidities. Out of 49 shared DEGs, most of the genes were upregulated while very few were downregulated (i.e. CXCL14, CYP4F3, MAP7D2, METTL7A, NANOS1, PPARGC1A and VTCN1). Interestingly, we found that COVID-19 shares most of the DEGs (28) with diabetes and 17 DEGs with LC. While the latter case is not surprising given the site of pathogenesis, such association with the former is quite interesting. It could be orchestrated by several genes acting in concert. For example, the upregulated IRF7 gene is associated with the pathogenesis of LC and type 1 diabetes [58, 59]. Further, the elevated expression of ADAM8 was observed in type 2 diabetes [60]. It also causes malignant cell growth and leads to poor outcomes in cancer patients [61]. The poor survival associated with ADAM8 expression is also supported by our survival analysis. In our study, ADAM8 was upregulated in COVID-19, DM and LC, and its upregulation revealed a poor prognosis in LC patients. Therefore, the results coincide with the previous study. Another gene MX2 encodes for interferon (IFN)-induced GTP-binding protein, which is found to be overexpressed in lung adenocarcinoma [62] and is involved in a better survival rate in skin cancer [63]. However, we did not find any significant difference in the survival outcome in LC with the expression of MX2 gene. COVID-19 shares the upregulation of CCL20 gene with LC and HT. It promotes LC through the proliferation and migration of cells using the PI3K and ERK signaling pathways [64]. Its overexpression is also involved in airway inflammation leading to severe asthma, HT, liver injury and LC [64–67]. Furthermore, we found upregulated CCL20 which is associated with better survival in LC patients. We observed that the upregulation of KRT6B is shared between COVID-19 and MI patients. Being a key mediator of the Notch signaling pathway, KRT6B enhances atherosclerosis by promoting endothelial dysfunction [68], which in turn may lead to the MI episode [69].
The manipulation and control of genes require their specific locations in the genome on which the expression depends. The majority of the shared DEGs are located at chromosomes 1, 11 and 12. Interestingly, in a study on a mouse model, chromosome 12 is found to be linked with airway hyper-responsiveness [70]. Besides, chromosome 11 holds fundamental genes related to metabolism and cell proliferation that are involved in a variety of cancers, especially in LC [71]. Moreover, most of the genes were found to be related to lung and spleen tissues. Therefore, our finding is supported by the characteristic features of COVID-19. To further validate the association of COVID-19 with selected comorbidities and other diseases, we applied the shared DEGs, which revealed potential risk factors and diseases. In addition to the selected comorbidities, the Metascape server suggested the top 20 complications, including reperfusion injury, liver cirrhosis and injury, psoriasis, dermatitis, rheumatoid arthritis, inflammation, ulcerative colitis, hypersensitivity, cancers, etc. Interestingly, recent reports are in line with our findings. For instance, the COVID-19 severity is prevalent in patients with liver cirrhosis and associated injuries [72, 73]. Studies suggested that psoriatic patients could be more susceptible to COVID-19 [74, 75]. Kutlu and Metin [76] recently reported that 9.6% of COVID-19 patients had a history of psoriasis. This is plausible since psoriasis developed due to the hyper-inflammatory reactions, which are highly prevalent in COVID-19 [74, 75]. Further, the shared DEGs were submitted in the Expression Atlas to determine their expression levels in associated comorbidities. Like in COVID-19, most of the shared DEGs were highly upregulated in various identified diseases, i.e. skin diseases, glioblastoma, psoriasis, Crohn disease, ulcerative disease, pneumonia and keratosis. Therefore, this finding coincides with the prediction done by the Metascape server, hence, validates our study as well.
Further, we selected the top 30 pathways that may play a crucial role in disease development. Notably, most of the genes and identified pathways are related to pro-inflammatory reactions (i.e. cytokine productions) and response to viral infections, which explains the cytokine storms in COVID-19 severity [16, 19]. The predicted pathways include Th1-secreted cytokines (i.e. lFN-γ, IL-1 and IL-2), Th2-mediated cytokines (i.e. IL-4, IL-13 and IL-10), IL-17 and tumor necrosis factor alpha (TNF-α) signaling pathways which cause widespread damage, respiratory failure and mortality [77, 78]. Interestingly, pro-inflammatory cytokines, especially Th1 cytokines, are known to increase insulin resistance [79, 80] that may facilitate additional complexities in a COVID-19 patient. Increased cytokine production is also associated with HT [81]. The preexisting cytokine imbalance in HT could make COVID-19 even worse and vice versa. Therefore, these pathways could be linked to COVID-19 and may reveal crucial checkpoints for drug targets. Due to the high prevalence of IL-17 in COVID-19, for instance, anti-IL-17 therapy has been considered as a potential treatment for COVID-19 [82]. Like pathways, GO terms pathways included the immune system process, defense response, cell surface receptor signaling, etc., which are relevant to SARS-CoV-2 infection.
Using the common DEGs, a PPI network was constructed to visualize their association and to determine the key disease-modifying players (hubs) in COVID-19 and comorbidities. Hubs are defined as proteins with eight or more interactions, while proteins with less than four interactions are named non-hubs [83]. Since they have many interacting partners within a network, hub-proteins are considered as functionally significant [84]. Using different methods, we identified six common hub-proteins (i.e. SOCS3, TLR2, BIRC3, PPARGC1A, HELZ2 and IRF7) involved in SARS-CoV-2 infection and the risk factors related to COVID-19. In our previous study, we identified SOCS3 as a hub-protein and observed its upregulation in SARS-like viral infections [32]. Another hub-protein BIRC3 is an apoptotic inhibitor and plays a crucial role in regulating NF-κB signaling and apoptosis [51]. It facilitates the malignant transformation of gliomas, a type of brain tumor, which is also evident in our study [52]. Therefore, the upregulation of BIRC3 due to COVID-19 may also initiate the progression of LC. Furthermore, the expression of SOCS3 downregulated the JAK2/STAT3 pathway to promote macrophage polarization that plays a key role in lung inflammation [47]. A previous study also identified MMP9 as an important biomarker in SARS-CoV-2 infection and respiratory failure [85]. Interestingly, MMP9 is also suggested as a hub-protein by several methods used in this study. This protein is well known for its involvement in both acute and chronic lung diseases [86]. In acute lung injury, MMP9 degrades cell matrices in lung tissues through neutrophil-mediated inflammation and destruction of the alveolar–capillary barrier [85, 87]. Therefore, these hub-proteins can be regarded as candidate biomarkers or, if their biological role in COVID-19 is confirmed, as potential drug targets. However, hub-proteins detected by the MNC method are completely different from other methods, which is conceivable since MNC scores the network differently [43].
Furthermore, TF controls the rate of transcription [88], while miRNA is a key player in RNA silencing and regulation of gene expression at the post-transcription level [89]. Hence, both are essential to understand particular disease development. This study unfolded relationships among the shared DEGs and their respective TFs and regulatory miRNAs. We identified several TFs, such as NFKB1A, E2F1, TP53 and CREB1, which are known to involve in viral-mediated acute respiratory diseases [90–92]. Among the 45 miRNAs, 10 were related to LC (i.e. miR-9-5p, 98-5p, 34a-5p, 30a-5p, 21-5p, 155-5p, 19a-3p, 19b-3p, 132-3p and 124-3p). The epithelial–mesenchymal transition (EMT) is crucial for the invasion, migration and metastasis of cancers, which is regulated by miR-19a-3p and 19b-3p [93]. The miR-155-5p controls the apoptosis and deoxyribonucleic acid (DNA) damage using the Apaf-1-mediated pathway [94]. Again, miR-21-5p is upregulated in non-small LC, while its downregulation reduces cell proliferation [93]. Likewise, miR-21-5p, 34a-5p and 143-3p are found to be associated with different cardiovascular diseases, including stroke, heart failure, coronary artery disease and HT [95, 96]. Specifically, miR-21 involved in cardiac remodeling by targeting the ERK/MAP pathway, while miR-143 targets NF-κB and ACE-related pathways for macrophage differentiation and polarization [95]. The presence of miR-122 is observed to be upregulated briefly after the initiation of ischemia. Further, miR-125b-5p is upregulated in DM patients and is involved in insulin resistance as well as the immune response to viral infections [97].
In DM patients, miR-30c is downregulated and it increases the activation of P53 pathway and apoptosis [98]. Conversely, upregulated miR-34a-5p increases high glucose-mediated apoptosis in cardiomyocytes by reducing anti-apoptotic BCL2 protein [99]. Thus, the upregulation of miR-34a-5p in the diabetic condition is most likely to increase apoptosis [98]. Interestingly, the elevated level of miRNA-34a was also observed in patients who experience acute MI leading to heart failure [98]. Moreover, some miRNAs associated with asthma (i.e. miR-155-5p) and pneumonia (i.e. miR-30a-5p) were also found, which are highly prevalent in COVID-19 [92, 100, 101]. Overall, these data and those included in the respective supplementary file(s) are of clinical interest and may shed light on the cause and progression of COVID-19 as well as any new prospective therapeutic strategies. By considering the common DEGs, several drug candidates are also proposed. As expected, several drugs have already been found to have therapeutic effects against the SARS-CoV-2 infection. For example, pre-treatment with captopril, an angiotensin-converting enzyme inhibitor (ACEI), has shown to reduce the level of ACE2 and promote anti-inflammatory reactions that may contribute to the better outcome of the COVID-19 patients [102]. We also identified rilonacept which is an approved IL-1 inhibitor. In a recent study, mortality risk from COVID-19 was significantly reduced among the patients treated with IL-1 inhibitor [103], hence, rilonacept should further be assessed for its anti-COVID-19 action. Another identified drug is canakinumab, which restored normal oxygen level in patients suffering from COVID-19-related pneumonia [104]. Therefore, the identified drug candidates should be evaluated for their protective effect on COVID-19 patients.
Finally, we evaluated the effect of COVID-19 on the prognostic outcome of LC patients. Importantly, the expression pattern of most DEGs in COVID-19 was found to be associated with poor survival outcomes in patients with LC. In earlier studies, for instance, downregulation of METTL7A and upregulation of CXCL14 had been found to be associated with the worst OS [105, 106]. Most crucially, CXCL14 was reported to be involved in the progression of LC from chronic obstructive pulmonary disease (COPD) [106]. Therefore, it may also be a key determinant in transforming the COVID-19 into LC metastasis. We observed that the PPARGC1A is downregulated in COVID-19 and is associated with a poor LC prognosis. Further, we identified PPARGC1A as a hub-protein in COVID-19 and LC. A previous study suggests PPARGC1A as a potential biomarker in LC prognosis [54]. Therefore, other identified hubs could also be potential biomarkers in COVID-19 and LC prognosis. However, unlike in this study, the upregulation of PPARGC1A facilitates LC metastasis [54]. Likewise, COVID-19 results in IRF7 overexpression, which was subsequently found to deteriorate the LC prognosis. Nevertheless, an early study suggested that the silencing of IRF7 may increase the virus-mediated killing of LC cells, therefore, better survival outcome [58]. Furthermore, IFITM1 is upregulated in COVID-19 and LC metastasis [107]. Proliferation, migration and invasion in LC can be inhibited by silencing the IFITM1 gene suggested in a previous in vitro study [107]. Therefore, genes that are related to poor LC outcomes can be targeted to hinder/stop the development of LC following the COVID-19 as well as to ameliorate the OS outcome.
Conclusion
To sum up, we compared the gene expression profile of COVID-19 with associated four comorbidities to understand the possible synergistic outcome. Using the network-based approach, we identified important proteins, regulatory molecules, signaling pathways and gene ontologies as well as other potential risk factors. While these networks may reveal important disease-modifying elements as potential drug targets, the signaling pathways can unfold important molecular checkpoints for the development of novel therapeutics in COVID-19 eradication. The identified biomarkers can be utilized to design new diagnostic tools and as drug targets based on their role in the progression of diseases. Furthermore, as suggested by the prognostic analysis, poor survival outcome in LC patients may result due to the altered gene expression caused by SARS-CoV infection. These genes can be silenced to inhibit the LC progression and to ameliorate the overall prognosis. Nevertheless, the feasibility to use and/or target the genetic determinants in fighting the pandemic requires further experimental validation.
Key Points
This transcriptome-based cross-comparative analysis unfolded genetic determinants causing severity and progression of COVID-19 in patients with predominant diseases.
The PPI network revealed important hub-proteins that might play key roles in disease development and can be evaluated for prognostic biomarkers.
Gene ontological groups and signaling pathways provided information about the processes that are involved in pathogenic progression and their implication in prevalent diseases.
The regulatory molecules identified in gene–miRNA and gene–TF networks could improve our understanding of the disease development and can be used as a possible drug target.
Protein–drug interactome suggests potential drugs molecules that should be evaluated for their protective actions in COVID-19 and associated complications.
Supplementary Material
Zulkar Nain is a research assistant at the Department of Biotechnology and Genetic Engineering, Islamic University, Bangladesh. His research interests are infection biology, functional genomics, molecular modeling and dynamics as well as translational bioinformatics.
Shital K. Barman is a PhD candidate at Western Sydney University, Australia. He obtained his MSc and BSc degrees from Islamic University, Bangladesh. He is currently involved in researches focused on breast and prostate cancers.
Md. Moinuddin Sheam has graduated from the Department of Biotechnology and Genetic Engineering, Islamic University, Bangladesh, and is working as a research assistant at the same department. He endures interest in computational biology, bioremediation and environmental and infection microbiology.
Shifath Bin Syed is a research assistant at the Department of Biotechnology and Genetic Engineering, Islamic University, Bangladesh. He possesses a keen interest in computational biology, antibiotic resistance, infection microbiology and microbial pathogenesis.
Abdus Samad is an undergraduate student at the Department of Genetic Engineering and Biotechnology at the Jashore University of Science and Technology, Bangladesh. His research interest includes gene expression analysis, biomarker discovery and pharmaceutical biotechnology.
Julian M. W. Quinn is a senior research officer at the Garvan Institute of Medical Research, Australia. He received his PhD from the University of Oxford and postdoctoral training in bone and cancer biology at the Melbourne University and St Vincent’s Institute of Medical Research.
Mohammad Minnatul Karim is a professor at the Islamic University, Bangladesh. He obtained his PhD degree from the Tottori University, Japan. His research interest broadly lies in quorum sensing, biofilm development, antibiotic resistance and computational biology.
Mahbubul Kabir Himel is an associate professor at the Jahangirnagar University, Bangladesh. His research focus includes antibiotic resistance, structural studies of β-lactamase and drug discovery.
Rajib Kanti Roy is an assistant professor at the Jashore University of Science and Technology, Bangladesh. He completed his BSc and MSc degrees in Food and Nutrition Technology, and his research interests include molecular nutrition, diabetes and computational biology.
Mohammad Ali Moni is a research fellow and conjoint lecturer at the University of New South Wales, Australia. He received his PhD degree from the University of Cambridge. His research interest encompasses artificial intelligence, machine learning, data science and clinical bioinformatics.
Sudhangshu Kumar Biswas is an associate professor at the Islamic University, Bangladesh. He obtained his PhD degree from the University of Malaya, Malaysia. His research interest broadly lies in bacteriophages, phage therapy, bioremediation, genomics and bioinformatics.
Contributor Information
Zulkar Nain, Department of Biotechnology and Genetic Engineering, Islamic University, Bangladesh.
Shital K Barman, Islamic University, Bangladesh.
Md Moinuddin Sheam, Department of Biotechnology and Genetic Engineering, Islamic University, Bangladesh.
Shifath Bin Syed, Department of Biotechnology and Genetic Engineering, Islamic University, Bangladesh.
Abdus Samad, Department of Genetic Engineering and Biotechnology at the Jashore University of Science and Technology, Bangladesh.
Julian M W Quinn, Garvan Institute of Medical Research, Australia.
Mohammad Minnatul Karim, Islamic University, Bangladesh.
Mahbubul Kabir Himel, Jahangirnagar University, Bangladesh.
Rajib Kanti Roy, Jashore University of Science and Technology, Bangladesh.
Mohammad Ali Moni, University of New South Wales, Australia.
Sudhangshu Kumar Biswas, Islamic University, Bangladesh.
Data availability
All the supplementary data supporting the analysis and interpretation of results presented in this study are available in Harvard Dataverse (‘Supplementary DATA: Transcriptomic Studies Revealed Pathophysiological Impact of COVID-19 to Predominant Health Conditions’, https://doi.org/10.7910/DVN/8JEUAN, Harvard Dataverse, V1). Data are available under the terms of the Creative Commons Zero ‘No rights reserved’ data waiver (CC0 1.0 Public domain dedication).
Authors’ contributions
M.A.M. and Z.N. conceived and designed the study; Z.N., S.B.S. and M.M.S. performed most of the computational analyses; Z.N., A.S. and M.A.M. executed the survival analysis and interpretation; Z.N. prepared the illustrations and wrote the manuscript; Sh.K.B., M.K.H. and R.K.R. helped in the experimental procedure and draft preparation; Su.K.B., M.A.M., J.M.W.Q. and M.M.K. critically reviewed the manuscript and provided important suggestions; M.A.M. and Su.K.B. supervised the whole study. All authors approved the final version of the manuscript.
Funding statement
This research received no external grant from any funding agency in the public, commercial, or non-profit sectors.
Ethical statement
Not applicable.
References
- 1. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 2020;382:727–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sheam M, Syed S, Barman S, et al. COVID-19: the catastrophe of our time. J Adv Biotechnol Exp Ther 2020;3(4):1–13. [Google Scholar]
- 3. Sharma A, Tiwari S, Deb MK, et al. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): a global pandemic and treatment strategies. Int J Antimicrob Agents 2020;56(2):106054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu D, Wu T, Liu Q, et al. The SARS-CoV-2 outbreak: what we know. Int J Infect Dis 2020;94:44–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China Lancet 2020;395(10223):497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abu Hammad O, Alnazzawi A, Borzangy SS, et al. Factors influencing global variations in COVID-19 cases and fatalities; a review. Dent Health 2020;8(3):216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lakshmi Priyadarsini S, Suresh M. Factors influencing the epidemiological characteristics of pandemic COVID 19: a TISM approach. Int J Healthc Manag 2020;13(2):89–98. [Google Scholar]
- 8. Bajgain KT, Badal S, Bajgain BB, et al. Prevalence of comorbidities among individuals with COVID-19: a rapid review of current literature. Am J Infect Control 2020;49(2):238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. fen Zhang S, ling Tuo J, bin Huang X, et al. Epidemiology characteristics of human coronaviruses in patients with respiratory infection symptoms and phylogenetic analysis of HCoV-OC43 during 2010-2015 in Guangzhou. PLoS One 2018;13(1):e0191789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sanyaolu A, Okorie C, Marinkovic A, et al. Comorbidity and its impact on patients with COVID-19. SN Compr Clin Med 2020;1:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 2020;395:1054–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Valderas JM, Starfield B, Sibbald B, et al. Defining comorbidity: implications for understanding health and health services. Ann Fam Med 2009;7(4):357–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moni MA, Lio P. Genetic profiling and comorbidities of Zika infection. J Infect Dis 2017;216(6):703–712. [DOI] [PubMed] [Google Scholar]
- 14. Wu C, Chen X, Cai Y, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med 2020;180(7):934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rodriguez-Morales AJ, Cardona-Ospina JA, Gutiérrez-Ocampo E, et al. Clinical, laboratory and imaging features of COVID-19: a systematic review and meta-analysis. Travel Med Infect Dis 2020;34:101623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Astuti I, Ysrafil. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): an overview of viral structure and host response. Diabetes Metab Syndr Clin Res Rev 2020;14:407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li X, Geng M, Peng Y, et al. Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal 2020;10:102–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mason RJ. Pathogenesis of COVID-19 from a cell biology perspective. Eur Respir J 2020;55:2000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rubino F, Amiel SA, Zimmet P, et al. New-onset diabetes in Covid-19. N Engl J Med 2020;383:789–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guan W, Liang W, Zhao Y, et al. Comorbidity and its impact on 1590 patients with Covid-19 in China: a nationwide analysis. Eur Respir J 2020;55(5):2000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morra ME, van Thanh L, Kamel MG, et al. Clinical outcomes of current medical approaches for Middle East respiratory syndrome: a systematic review and meta-analysis. Rev Med Virol 2018;28:e1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Furuhashi M, Moniwa N, Mita T, et al. Urinary angiotensin-converting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. Am J Hypertens 2015;28:15–21. [DOI] [PubMed] [Google Scholar]
- 23. Gupta R, Hussain A, Misra A. Diabetes and COVID-19: evidence, current status and unanswered research questions. Eur J Clin Nutr 2020;74:864–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guan W, Ni Z, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 2020;58(4):711–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Orioli L, Hermans MP, Thissen JP, et al. COVID-19 in diabetic patients: related risks and specifics of management. Ann Endocrinol (Paris) 2020;81(2):101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xue T, Li Q, Zhang Q, et al. Blood glucose levels in elderly subjects with type 2 diabetes during COVID-19 outbreak: a retrospective study in a single Center. medRxiv 2020; doi: 10.1101/2020.03.31.20048579. [DOI] [Google Scholar]
- 27. Apicella M, Campopiano MC, Mantuano M, et al. COVID-19 in people with diabetes: understanding the reasons for worse outcomes. Lancet. Diabetes Endocrinol 2020;8(9):782–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hamming I, Timens W, Bulthuis MLC, et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 2004;203:631–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cortiula F, Pettke A, Bartoletti M, et al. Managing COVID-19 in the oncology clinic and avoiding the distraction effect. Ann Oncol 2020;31(5):553–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luo J, Rizvi H, Preeshagul IR, et al. COVID-19 in patients with lung cancer. Ann Oncol 2020;31(10):1386–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rogado J, Pangua C, Serrano-Montero G, et al. Covid-19 and lung cancer: a greater fatality rate? Lung Cancer 2020;146:19–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nain Z, Rana HK, Liò P, et al. Pathogenetic profiling of COVID-19 and SARS-like viruses. Brief. Bioinformatics 2020;2020:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mura M, Cecchini MJ, Joseph M, et al. Osteopontin lung gene expression is a marker of disease severity in pulmonary arterial hypertension. Respirology 2019;24:1104–1110. [DOI] [PubMed] [Google Scholar]
- 34. Jiang N, Zou C, Zhu Y, et al. HIF-1α-regulated miR-1275 maintains stem cell-like phenotypes and promotes the progression of LUAD by simultaneously activating Wnt/β-catenin and Notch signaling. Theranostics 2020;10:2553–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Luo L, Zhou WH, Cai JJ, et al. Gene expression profiling identifies downregulation of the neurotrophin-MAPK signaling pathway in female diabetic peripheral neuropathy patients. J Diabetes Res 2017;2017:8103904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blanco-Melo D, Nilsson-Payant BE, Liu W-C, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020;181:1036–45.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shannon P. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ge SX, Jung D, Jung D, et al. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020;36:2628–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 2019;10:1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Papatheodorou I, Fonseca NA, Keays M, et al. Expression atlas: gene and protein expression across multiple studies and organisms. Nucleic Acids Res 2018;46(D1):D246–D251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44:W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou G, Soufan O, Ewald J, et al. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res 2019;47(W1):W234–W241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chin CH, Chen SH, Wu HH, et al. Cyto Hubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol 2014; 8 Suppl 4:S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Khan A, Fornes O, Stigliani A, et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 2018;46(D1):D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Da HS, Lin FM, Wu WY, et al. MiRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res 2011;39(Database issue):D163–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mizuno H, Kitada K, Nakai K, et al. Progno scan: a new database for meta-analysis of the prognostic value of genes. BMC Med Genomics 2009;2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chi X, Ding B, Zhang L, et al. Lnc RNA GAS5 promotes M1 macrophage polarization via miR-455-5p/SOCS3 pathway in childhood pneumonia. J Cell Physiol 2019;234:13242–51. [DOI] [PubMed] [Google Scholar]
- 48. Filippi CM, Ehrhardt K, Estes EA, et al. TLR2 signaling improves immunoregulation to prevent type 1 diabetes. Eur J Immunol 2011;41:1399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li K, Hao Z, Zhao X, et al. SARS-CoV-2 infection-induced immune responses: friends or foes? Scand J Immunol 2020;92(2):e12895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang D, Berglund A, Kenchappa RS, et al. BIRC3 is a novel driver of therapeutic resistance in glioblastoma. Sci Rep 2016;6:21710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yamato A, Soda M, Ueno T, et al. Oncogenic activity of BIRC2 and BIRC3 mutants independent of nuclear factor-κB-activating potential. Cancer Sci 2015;106(9):1137–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gressot LV, Doucette T, Yang Y, et al. Analysis of the inhibitors of apoptosis identifies BIRC3 as a facilitator of malignant progression in glioma. Oncotarget 2017;8:12695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rojek A, Cielecka-Prynda M, Przewlocka-Kosmala M, et al. Impact of the PPARGC1A Gly 482Ser polymorphism on left ventricular structural and functional abnormalities in patients with hypertension. J Hum Hypertens 2014;28:557–563. [DOI] [PubMed] [Google Scholar]
- 54. dong Li J, chuan Feng Q, Qi Y, et al. PPARGC1A is upregulated and facilitates lung cancer metastasis. Exp Cell Res 2017;359:356–60. [DOI] [PubMed] [Google Scholar]
- 55. Fusco DN, Pratt H, Kandilas S, et al. HELZ2 is an IFN effector mediating suppression of dengue virus. Front Microbiol 2017;8:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yoshino S, Satoh T, Yamada M, et al. Protection against high-fat diet-induced obesity in Helz2-deficient male mice due to enhanced expression of hepatic leptin receptor. Endocrinology 2014;155(9):3459–3472. [DOI] [PubMed] [Google Scholar]
- 57. Yuan L, Chen Z, Song S, et al. P53 degradation by a coronavirus papain-like protease suppresses type I interferon signaling. J Biol Chem 2015;290(5):3172–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li Q, Tainsky MA. Epigenetic silencing of IRF7 and/or IRF5 in lung cancer cells leads to increased sensitivity to oncolytic viruses. PLoS One 2011;6:28683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ning S, Pagano JS, Barber GN. IRF7: activation, regulation, modification and function. Genes Immun 2011;12(6):399–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Worley JR, Hughes DA, Dozio N, et al. Low density lipoprotein from patients with type 2 diabetes increases expression of monocyte matrix metalloproteinase and ADAM metalloproteinase genes. Cardiovasc Diabetol 2007;6:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Conrad C, Benzel J, Dorzweiler K, et al. ADAM8 in invasive cancers: links to tumor progression, metastasis, and chemoresistance. Clin Sci 2019;133(1):83–99. [DOI] [PubMed] [Google Scholar]
- 62. Kobayashi K, Nishioka M, Kohno T, et al. Identification of genes whose expression is upregulated in lung adenocarcinoma cells in comparison with type II alveolar cells and bronchiolar epithelial cells in vivo. Oncogene 2004;23:3089–96. [DOI] [PubMed] [Google Scholar]
- 63. Juraleviciute M, Pozniak J, Nsengimana J, et al. MX 2 is a novel regulator of cell cycle in melanoma cells. Pigment Cell Melanoma Res 2020;33(3):446–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang HF, Chen L, Luo J, et al. KLF5 is involved in regulation of IFITM1, 2, and 3 genes during H5N1 virus infection in A549 cells. Cell Mol Biol 2016;62(13):65–70. [DOI] [PubMed] [Google Scholar]
- 65. Nozato K, Fujita J, Kawaguchi M, et al. IL-17F induces CCL20 in bronchial epithelial cells. J Allergy 2011;2011:587204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang XP, Hu ZJ, Meng AH, et al. Role of CCL20/CCR6 and the ERK signaling pathway in lung adenocarcinoma. Oncol Lett 2017;14:8183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Affò S, Morales-Ibanez O, Rodrigo-Torres D, et al. CCL20 mediates lipopolysaccharide induced liver injury and is a potential driver of inflammation and fibrosis in alcoholic hepatitis. Gut 2014;63(11):1782–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pan Y, Wang L, Xie Y, et al. Characterization of differentially expressed plasma proteins in patients with acute myocardial infarction. J Proteomics 2020;227:103923. [DOI] [PubMed] [Google Scholar]
- 69. Dutta P, Courties G, Wei Y, et al. Myocardial infarction accelerates atherosclerosis. Nature 2012;487:325–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kanagaratham C, Marino R, Camateros P, et al. Mapping of a chromosome 12 region associated with airway hyperresponsiveness in a recombinant congenic mouse strain and selection of potential candidate genes by expression and sequence variation analyses. PLoS One 2014;9:e104234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pitterle D, Kim Y-C, Cao Y, et al. The pathogenesis of lung cancer and chromosome 11. Curr Genomics 2000;1:281–8. [Google Scholar]
- 72. Moon AM, Webb GJ, Aloman C, et al. High mortality rates for SARS-CoV-2 infection in patients with pre-existing chronic liver disease and cirrhosis: preliminary results from an international registry. J Hepatol 2020;73:705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pawlotsky J-M. COVID-19 and the liver-related deaths to come. Nat Rev Gastroenterol Hepatol 2020;17:523–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Elmas ÖF, Demirbaş A, Kutlu Ö, et al. Psoriasis and COVID−19: a narrative review with treatment considerations. Dermatol Ther 2020;33(6):e13858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ebrahimi A, Sayad B, Rahimi Z. COVID-19 and psoriasis: biologic treatment and challenges. J Dermatolog Treat 2020;1–5. [DOI] [PubMed] [Google Scholar]
- 76. Kutlu Ö, Metin A. Dermatological diseases presented before COVID-19: are patients with psoriasis and superficial fungal infections more vulnerable to the COVID-19? Dermatol Ther 2020;33(4):e13509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schett G, Manger B, Simon D, et al. COVID-19 revisiting inflammatory pathways of arthritis. Nat Rev Rheumatol 2020;16:465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Harrison C. Focus shifts to antibody cocktails for COVID-19 cytokine storm. Nat Biotechnol 2020;38:905–8. [DOI] [PubMed] [Google Scholar]
- 79. Lee B-C, Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim Biophys Acta-Mol Basis Dis 2014;1842:446–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lim S, Bae JH, Kwon H-S, et al. COVID-19 and diabetes mellitus: from pathophysiology to clinical management. Nat Rev Endocrinol 2021;17:11–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Drummond GR, Vinh A, Guzik TJ, et al. Immune mechanisms of hypertension. Nat Rev Immunol 2019;19:517–532. [DOI] [PubMed] [Google Scholar]
- 82. Ayhan E, Öztürk M, An İ, et al. Potential role of anti-interleukin-17 in COVID-19 treatment. Dermatol Ther 2020;33(4):e13715. [DOI] [PubMed] [Google Scholar]
- 83. Ekman D, Light S, Björklund ÅK, et al. What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae? Genome Biol 2006;7:R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. He X, Zhang J. Why do hubs tend to be essential in protein networks? PLoS Genet 2006;2:e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hazra S, Chaudhuri AG, Tiwary BK, et al. Matrix metallopeptidase 9 as a host protein target of chloroquine and melatonin for immunoregulation in COVID-19: a network-based meta-analysis. Life Sci 2020;257:118096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Davey A, McAuley DF, O’Kane CM. Matrix metalloproteinases in acute lung injury: mediators of injury and drivers of repair. Eur Respir J 2011;38(4):959–970. [DOI] [PubMed] [Google Scholar]
- 87. Ueland T, Holter JC, Holten AR, et al. Distinct and early increase in circulating MMP-9 in COVID-19 patients with respiratory failure. J Infect 2020;81(3):e41–e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Latchman DS. Transcription factors: an overview. Int J Biochem Cell Biol 1997;29:1305–12. [DOI] [PubMed] [Google Scholar]
- 89. Ambros V. The functions of animal microRNAs. Nature 2004;431:350–355. [DOI] [PubMed] [Google Scholar]
- 90. Bartel S, Schulz N, Alessandrini F, et al. Pulmonary microRNA profiles identify involvement of Creb1 and Sec14l3 in bronchial epithelial changes in allergic asthma. Sci Rep 2017;7:46026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Krug N, Hohlfeld JM, Kirsten AM, et al. Allergen-induced asthmatic responses modified by a GATA3-specific DNAzyme. N Engl J Med 2015;372(21):1987–1995. [DOI] [PubMed] [Google Scholar]
- 92. Leon-Icaza SA, Zeng M, Rosas-Taraco AG. microRNAs in viral acute respiratory infections: immune regulation, biomarkers, therapy, and vaccines. ExRNA 2019;1:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wu SG, Chang TH, Liu YN, et al. Micro RNA in lung cancer metastasis. Cancers (Basel) 2019;11(2):265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Castro D, Moreira M, Gouveia AM, et al. Micro RNAs in lung cancer. Oncotarget 2017;8:81679–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ultimo S, Zauli G, Martelli AM, et al. Cardiovascular disease-related miRNAs expression: potential role as biomarkers and effects of training exercise. Oncotarget 2018;9:17238–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Peng L, Ma W, Yi F, et al. Micro RNA profiling in Chinese patients with primary Sjögren syndrome reveals elevated miRNA-181a in peripheral blood mononuclear cells. J Rheumatol 2014;41:2208–13. [DOI] [PubMed] [Google Scholar]
- 97. Satake E, Pezzolesi MG, Md Dom ZI, et al. Circulating miRNA profiles associated with hyperglycemia in patients with type 1 diabetes. Diabetes 2018;67:1013–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hathaway QA, Pinti MV, Durr AJ, et al. Regulating microRNA expression: at the heart of diabetes mellitus and the mitochondrion. Am J Physiol Circ Physiol 2018;314:H293–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhao F, Li B, Wei YZ, et al. Micro RNA-34a regulates high glucose-induced apoptosis in H9c2 cardiomyocytes. J Huazhong Univ Sci Technolog Med Sci 2013;33(6):834–839. [DOI] [PubMed] [Google Scholar]
- 100. Herrera-Rivero M, Zhang R, Heilmann-Heimbach S, et al. Circulating microRNAs are associated with pulmonary hypertension and development of chronic lung disease in congenital diaphragmatic hernia. Sci Rep 2018;8:10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kho AT, Sharma S, Davis JS, et al. Circulating micro RNAs: association with lung function in asthma. PLoS One 2016;11(6):e0157998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Pedrosa MA, Valenzuela R, Garrido-Gil P, et al. Experimental data using candesartan and captopril indicate no double-edged sword effect in COVID-19. Clin Sci 2021;135:465–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Cavalli G, Larcher A, Tomelleri A, et al. Interleukin-1 and interleukin-6 inhibition compared with standard management in patients with COVID-19 and hyperinflammation: a cohort study. Lancet Rheumatol 2021;3(4):e253–e261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Generali D, Bosio G, Malberti F, et al. Canakinumab as treatment for COVID-19-related pneumonia: a prospective case-control study. Int J Infect Dis 2021;104:433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Guo T, Ma H, Zhou Y. Bioinformatics analysis of microarray data to identify the candidate biomarkers of lung adenocarcinoma. Peer J 2019;7:e7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Shaykhiev R, Sackrowitz R, Fukui T, et al. Smoking-induced CXCL14 expression in the human airway epithelium links chronic obstructive pulmonary disease to lung cancer. Am J Respir Cell Mol Biol 2013;49:418–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yang YG, Koh YW, Sari IN, et al. Interferon-induced transmembrane protein 1-mediated EGFR/SOX2 signaling axis is essential for progression of non-small cell lung cancer. Int J Cancer 2019;144:2020–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the supplementary data supporting the analysis and interpretation of results presented in this study are available in Harvard Dataverse (‘Supplementary DATA: Transcriptomic Studies Revealed Pathophysiological Impact of COVID-19 to Predominant Health Conditions’, https://doi.org/10.7910/DVN/8JEUAN, Harvard Dataverse, V1). Data are available under the terms of the Creative Commons Zero ‘No rights reserved’ data waiver (CC0 1.0 Public domain dedication).