

Abstract
Coronavirus disease 2019 pandemic is the most damaging pandemic in recent human history. Rapid detection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and variant strains is paramount for recovery from this pandemic. Conventional SARS-CoV-2 tests interrogate only limited regions of the whole SARS-CoV-2 genome, which are subjected to low specificity and miss the opportunity of detecting variant strains. In this work, we developed the first SARS-CoV-2 tiling array that captures the entire SARS-CoV-2 genome at single nucleotide resolution and offers the opportunity to detect point mutations. A thorough bioinformatics protocol of two base calling methods has been developed to accompany this array. To demonstrate the effectiveness of the tiling array, we genotyped all genomic positions of eight SARS-CoV-2 samples. Using high-throughput sequencing as the benchmark, we show that the tiling array had a genome-wide accuracy of at least 99.5%. From the tiling array analysis results, we identified the D614G mutation in the spike protein in four of the eight samples, suggesting the widespread distribution of this variant at the early stage of the outbreak in the United States. Two additional nonsynonymous mutations were identified in one sample in the nucleocapsid protein (P13L and S197L), which may complicate future vaccine development. With around $5 per array, supreme accuracy, and an ultrafast bioinformatics protocol, the SARS-CoV-2 tiling array makes an invaluable toolkit for combating current and future pandemics. Our SARS-CoV-2 tilting array is currently utilized by Molecular Vision, a CLIA-certified lab for SARS-CoV-2 diagnosis.
Keywords: SARS-CoV-2, COVID-19, tiling array
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the RNA virus that is responsible for the coronavirus disease 2019 (COVID-19) pandemic. As of February 2021, the world has suffered over 100 million infection cases, including over 2 million deaths from COVID-19. Currently, three distinct SARS-CoV-2 tests are used in clinical labs: molecular tests, antigen tests and antibody tests. Conducted through the authoritative RT-PCR technique, molecular tests seek to detect viral-specific nucleotide sequences of 100–300 base-pair (bp) long [1]. Molecular tests are considered the most sensitive of all types of detection methods, although presenting an issue of inevitably high false-positive rates [2, 3]. The second type of test, antigen tests, detects specific proteins on the viral surface. People testing positive for an antigen via this test are usually at the peak phase postinfection. Lastly, antibody tests look for antibodies directed to SARS-CoV-2. People who have contracted SARS-CoV-2 or received SARS-CoV-2 vaccination are most likely to test positive in an antibody test. A study [4] has shown low sensitivity for IgG/IgM-based antibody tests, urging the development of additional more accurate tests.
SARS-CoV-2 is an RNA virus that exploits all known biological mechanisms of genetic variation to ensure survival. High mutation rate is one of the distinctive features of RNA viruses [5], providing an evolutionary mechanism for increased survival and transmission. The mutation rate of RNA viruses is estimated up to one million times higher than their host [6]. Based on the quasispecies theory, a high mutation rate can be selectively advantageous for low population size and small genome RNA viruses [7]. While the majority of the mutated strains will not survive, those that do survive can be fatal to humans and can develop resistance to established treatments. In February 2020, 2 months after the initial outbreak, thousands of SARS-CoV-2 variants had already been identified [8]. While the majority of SARS-CoV-2 variants do not pose a new challenge, several variants have gained viable advantages. Notable SARS-CoV-2 variants include the 501.V1 variant, also known as B.1.1.7, which was first detected in the UK in October 2020, and the 501.V2 variant, also known as B.1.351, which was first detected in South Africa. Both variants differ from the original SARS-CoV-2 strain by an overtly large number of mutations; both variants exhibit unusually high transmissibility and possible antibody evasion [9].
SARS-CoV-2 accesses human cells through its spike proteins that are like tiny ‘clove’ like proteins protruding from the surface of the viral outer envelopes. The current vaccines (AstraZeneca, Johnson and Johnson, Pfizer and Moderna) simulate the spike proteins to trigger an immune system to generate proper protective antibodies. Excessive mutations in the spike protein could lead to variants that elude the immune system, thus impeding a spontaneous defensive response. Researchers are still racing to elucidate these SARS-CoV-2 variants and their impacts on the immune response system.
Microarray technology was the backbone for gene expression and genetic variant studies until high-throughput sequencing became mature and affordable. Despite fearsome competition from the sequencing technology, microarrays still bear many nonnegligible advantages and offer novel data mining opportunities [10]. Given the manageable size of the SARS-CoV-2 genome and the cost-efficiency and maturity of microarray technology, we developed a new tiling array technology to resequence the entire SARS-CoV-2 genome for the sake of detecting SARS-CoV-2 infection and identifying single nucleotide variants. Here, we describe the design of the SARS-CoV-2 tiling array and two companion bioinformatics algorithms to reveal the SARS-CoV-2 variant genome. The utility of the hardware and software was demonstrated on SARS-CoV-2 samples extracted from eight COVID-19 patients.
Methods
Sample collection
Eight deidentified SARS-CoV-2–positive clinical samples from the Wyoming Public Health Laboratory were obtained and used in this study. All samples were collected before May 2020. They are referred to as S1 to S8 in this study. No consent was necessary because only deidentified viral materials were used for this study; no human samples were collected or used.
Sample preparation for Illumina sequencing
Samples were prepared as previously described using the ARTIC sequencing methods. In brief, cDNA was prepared from total RNA extracted from clinical samples using SuperScript IV (SSIV, Thermo Scientific) and random hexamer priming. The resultant cDNA was amplified in two PCR reactions using the ARTIC Pool1 and Pool2 SARS-CoV-2 v3 primer sets and Q5 High-Fidelity DNA Polymerase (NEB). Following PCR, samples were purified using AMPure XP SPRI beads (Beckman Coulter). Illumina adaptors were added using the NEBNext® Ultra™ II DNA Library Prep Kit (NEB) and SPRI bead purification was repeated.
Illumina sequencing data processing
Sequencing data of SARS-CoV-2 were aligned using BWA 0.7.17 [11] using reference genome NC_045512 downloaded from NCBI. Binary alignment map (BAM) files were sorted and indexed using Samtools 1.9 [12]. BCFTools 1.9 was used to count allele frequency from the BAM files.
Sample preparation for chip hybridization
To prepare samples for hybridization to the chips, 0.05 μL of purified PCR product was reamplified using the ARTIC protocol and Pool1 and Pool2 v3 primer sets for 35 cycles with 50 μM biotin-11-dUTP (Jena Biosciences) added to the reaction mixture. Pool1 and Pool2 were combined for each sample and fragmented using DNase I (D4263, Sigma). About 2000 Kunitz units of lyophilized DNase I was resuspended on ice using 2 mL of 1x DNase I Buffer (10 mM Tris–HCl pH 7.5, 2.5 mM MgCl2, 0.1 mM CaCl2). The resuspended enzyme was diluted 1000-fold using 1x DNase I Buffer and an equal volume was added to samples prewarmed to 37°C. Samples were incubated for 30 min at 37°C and the reactions were stopped by adding EDTA to a final concentration of 12.5 mM and incubating for 20 min at 75°C.
Hybridization
About 45 μL of the fragmented sample was hybridized overnight at 45°C to the chip in a 60 μL final volume containing 5 mM EDTA, 6.25 mM HEPES pH 8.0, 312.5 mM NaCl, 1.25% FIcoll 400, 0.5 nM Cy3-AM1 (GCTGTATCGGCTGAATCGTA). Following hybridization, chips were washed for 10 min at room temperature in Wash A (2x SSC, 0.1% TWEEN-20) and then for 10 min at 39°C in Wash B (0.5xSSC, 0.1% TWEEN-20). Chips were stained for 15 min at room temperature using 0.02 mg/mL Cy3-Streptavidin (Thermo) in 4x SSC and washed for 5 min at room temperature using 4xSSC. Chips were scanned using a custom-built confocal scanner for 0.5, 1, 4 and 8 s in the green (Cy3) channel in 4x SSC.
SARS-CoV-2 tiling array design
A tiling array is based on traditional microarray technology but aimed to resequence the entire genome. Our tiling array was designed based on the SARS-CoV-2 genome (NC_045512), which contains ~30 K nucleotides. Each position in the SARS-CoV-2 genome is captured by eight probes, four for sense and four for antisense strands. The four probes for the sense or antisense can be considered as a probe set, compactly designated as a ‘probeset’ hereafter. A probe is a 25-mer synthetic oligonucleotide matching the SARS-CoV-2 genome. In each probeset, the only difference among the probes is the middle nucleotide, which is used to capture the four possible variants (A, T, C, G) at that specific position (Figure 1A). Sense and antisense probesets are designed based on the same concept except that the antisense probes are used to capture the same information from the antisense strand resulting from reverse transcribed double-strand cDNA. Because the probe design requires 12 flanking sequences from either side of the targeted base, the first and last 12 bases of SARS-CoV-2 genome are not covered by the tiling array. In total, there are 239 000 probes on the tiling array. Each probe occupies a 3μm2 area and the entire tiling array is 3mm2. Due to variations in the intensity within the array, we elected to scan the array with three different exposure times (0.5, 1 and 4 s). Longer exposure time allows more accurate intensity profiling on weak intensity regions. However, it also saturates the brighter regions on the array. A proper summation method is required to combine the results from the six replicates (two strands x three exposures) for a same genomic position.
Figure 1 .

The overall tiling array design and bioinformatics analysis strategy. (A) The array design overview. Each position of the SARS-CoV-2 genome is covered by two probesets (sense and antisense). Each probeset contains four probes; each probe is a 25mer synthetic oligonucleotide, with middle nucleotide trying to capture the actual allele from the sample. Three exposure times were used to scan the array. (B) Two bioinformatics approaches were developed for inferring the allele: weighted voting method and ranked credibility score method.
Base calling algorithms
Considering the strandness and the exposure parameter, each nucleotide position in the SARS-CoV-2 genome is represented with six probesets (two strands × three exposures) on the tiling array. Within a probeset, the sorted intensity values returned by individual probes are represented as  . The highest intensity
. The highest intensity  identifies a probe of a specific nucleotide base, one from
identifies a probe of a specific nucleotide base, one from  , and that specific nucleotide base is denoted as a tentative call from the probeset in question. To make an ultimate base call for each position in the genome, we developed two base calling methods: weighted voting and ranked credibility score (Figure 1B).
, and that specific nucleotide base is denoted as a tentative call from the probeset in question. To make an ultimate base call for each position in the genome, we developed two base calling methods: weighted voting and ranked credibility score (Figure 1B).
Weighted voting uses the following protocol
Recalling that all probesets of replicate series
 (a specific configuration of strand and exposure) represent all nucleotide positions of the whole genome, let Ni be the number of positions with tentative calls consistent with the reference genome. Across all six replicate series, let
(a specific configuration of strand and exposure) represent all nucleotide positions of the whole genome, let Ni be the number of positions with tentative calls consistent with the reference genome. Across all six replicate series, let  be the number of positions where all six tentative calls are in agreement and are consistent with the reference genome. Accordingly, each replicate series gives rise to a reliability value
be the number of positions where all six tentative calls are in agreement and are consistent with the reference genome. Accordingly, each replicate series gives rise to a reliability value  , and the six replicate series altogether present a reliability vector
, and the six replicate series altogether present a reliability vector  . The raw reliability vector is normalized to a weight vector
. The raw reliability vector is normalized to a weight vector  , with
, with  .
.For each probeset, the raw highest intensity (I3) is converted to relative highest intensity as
 . Thus, at a nucleotide position in the SARS-CoV-2 genome, the relative highest intensity values of six probesets form a vector
. Thus, at a nucleotide position in the SARS-CoV-2 genome, the relative highest intensity values of six probesets form a vector  .
.For each position in SARS-CoV-2 genome, the relative highest intensity vector (Iv) is multiplied by the reliability weight vector dimension-wise, leading to a decision-making vector
 . The circled-dot symbol (
. The circled-dot symbol ( ) here means element-wise multiplication.
) here means element-wise multiplication.For each candidate base
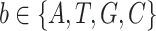 , identify the probesets whose tentative call points to it and sums up the corresponding dimension values from the decision-making vector V to yield base strength Sb. For example, if the six replicate series sequentially nominate A, T, A, G, C and G in their tentative calls, the base strengths for the four candidate bases are formulated as
, identify the probesets whose tentative call points to it and sums up the corresponding dimension values from the decision-making vector V to yield base strength Sb. For example, if the six replicate series sequentially nominate A, T, A, G, C and G in their tentative calls, the base strengths for the four candidate bases are formulated as  ,
,  ,
,  and
and  .
.The ultimate base call for the concerned genomic position is the nucleotide of the largest base strength, i.e.
 .
.
Ranked credibility score uses the following protocol
For each probeset, two dominance measures are assessed:
 and
and  . Both
. Both  and
and  measure how dominant the highest intensity (
measure how dominant the highest intensity ( ) is in relation to the other subordinate probes in the same probeset.
) is in relation to the other subordinate probes in the same probeset.Tentative base calls are first made for all probesets of all replicate series. By referring to the corresponding nucleotide position in the reference genome, the whole set of tentative base calls are classified into a Ref-agree group (consistent with the reference genome) and a Ref-nonagree group (inconsistent with the reference genome). The dominance measures for the two separate groups form four sets,
 and
and  .
.For one of the six probesets for a genomic position, the specific dominance measures (
 ,
, ) are converted to a credibility score Si (i = 1, 2, …, 6) by taking into account of the observed. Note that here
) are converted to a credibility score Si (i = 1, 2, …, 6) by taking into account of the observed. Note that here  , differentiating the six different replicate series.
, differentiating the six different replicate series.  , where the informal function notation #(condition) designates the number of elements meeting the specified condition.
, where the informal function notation #(condition) designates the number of elements meeting the specified condition.The most credible replicate series is identified by judging the credibility scores as
 ,
,  .
.The ultimate base call for the concerned genomic position is the tentative call of the replicate series i* that is associated with the maximum credibility score.
By default, the main results reported in this study were generated by the weighted voting method, unless otherwise stated explicitly.
Evaluation metrics with running genome windows
We calculated three metrics for a segment of nucleotide sequence along the SARS-CoV-2 genome. The GC content is dependent on the reference genome of SARS-CoV-2. The sequencing depth is dependent on the high-throughput sequencing result. The replicate consistency is defined specifically for the SARS-CoV-2 tiling array data. GC content is the percentage of guanine (G) and cytosine (C) bases within the nucleotide segment. Sequencing depth is the average reads coverage within the nucleotide segment. Across all segments of the whole genome, the sequencing depth values were standardized to a minimum of 0 and a maximum of 1. Replicate consistency is first defined for one exact nucleotide position as n/6 with n being the number of replicate probesets supporting the ultimate position base call; furthermore, replicate consistency of a nucleotide segment is obtained as the average number across all the individual positions within the segment. Given the linear single strand of SARS-CoV-2 genome, we typically binned the genome in running, nonoverlapping windows of 100 bp wide, unless specified otherwise. The aforementioned metrics, GC content, sequencing depth and replicate consistency, are calculated for each running bin and are thus connected into trend lines along the whole genome.
Results
Unanimity and consistency of base calls
The SARS-CoV-2 tiling array builds in six replicated intensity readings for each SARS-CoV-2 sample. For the eight samples we tested, the intraclass correlation coefficient [13] between the six replicate series ranged over 0.88–0.90 with an average of 0.89, and 79.8–87.1% (mean: 84.6%) of all 30 K genomic positions showed perfect replicate consistency, i.e. unanimous tentative calls among six replicates. For all genomic positions represented on a tiling array, we divided them into a probeset-unanimous group and a nonunanimous group. We calculated a noise statistics for each genomic position, which was defined as the reciprocal of relative highest intensity (see METHODS). Because SARS-CoV-2 is a haploid genome, theoretically, only one allele should be observed at a genomic position. Hence, within a probeset, we may assume only one probe represents the correct base, while the other three, summarized in the noise statistics, represent background noise. Aggregating all eight samples, we plotted the distribution of the noise statistics for the unanimous positions and the nonunanimous positions separately. To our expectation, genomic positions of unanimous tentative calls from replicate probesets demonstrate a strikingly lower noise level than positions of nonunanimous tentative calls (Figure 2A). In other words, unanimous genomic positions generally show low noise level, or striking predominant intensity level, within each probeset.
Figure 2 .

Noise level and consistency of base calls. (A) Distribution of noise level for the unanimous and nonunanimous base calls from titling array data of eight samples. The background noise is lower with the unanimous calls than with the nonunanimous calls. (B) Heatmap of replicate consistency values for binned segments of SARS-Cov-2 genome across eight samples. For visualization clarity, the whole genome was segmented into 30 bins each of 1000 bp long. (C) Replicate consistency and its relatedness with sequencing depth and GC content. Both tiling array and sequencing suffer deficiency around roughly same genomic regions.
In the landscape of replicate consistency along the whole genome, several notable genomic regions exhibited minor consistency drops (Figure 2B). One such region was located between position number 19 300 and number 19 550, where distinctively lower consistency was visually recognizable from the preceding and succeeding levels. Interestingly, the sequencing depth trend line displayed evident dips at roughly the same genomic regions (Figure 2C). After averaging over the eight samples, the replicate consistency and sequencing depth had a Pearson correlation coefficient of 0.32 (P < 0.0001). This result suggests that tiling array hybridization and high-throughput sequencing were faced with the same difficulty along the SARS-CoV-2 genome. It is well known that GC content is associated with sequencing depth [14]. We summarized GC content of SARS-CoV-2 genome in sliding windows of 100-bp width. The overall GC content of SARS-CoV-2 genome is 38.0%, oscillating between 20% and 56% in sliding windows. Using linear regression, we found that GC content can explain a small portion of the variation in sequencing depth (adjusted R2 = 0.036, P = 0.0006). Similarly, GC content was also associated with array consistency (adjusted R2 = 0.13, P < 0.0001). It seems that GC content partially explains the genome region difficulties encountered by both tiling array hybridization and high-throughput sequencing. However, there are yet other undiscovered factors affecting the resolution of particular regions of the SARS-CoV-2 genome.
Validation via high-throughput sequencing
To validate the performance of SARS-CoV-2 tiling array, we conducted Illumina high-throughput sequencing on all eight samples. Using sequencing results as the gold standard, the tiling array displayed remarkable accuracy. On average, the eight samples had an accuracy of 99.61% (range: 99.50–99.78%) for the weighted voting method and 99.57% (range: 99.35–99.81%) for the ranked credibility score method, respectively (Figure 3). Such high accuracy certifies the high reliability of SARS-CoV-2 tiling array coupled with the base calling methods. The two base calling methods performed very similarly. The weighted voting method had a slight advantage in six of the eight samples, while the ranked credibility score method outperformed in the rest of the two samples. The two methods make the final decision via different mechanisms: one by voting and the other by ranking, and they each may make correct calls in different scenarios. For example, in sample S2, at position number 22 908, the weighted voting method voted for nucleobase A based on the weighted decision-making vector of (1.073-A, 1.035-A, 1.027-A, 1.049-A, 1.0532-T and 1.060-A); the ranked credibility score method called nucleobase T based on the credibility score vector of (0.507-A, 0.156-A, 0.669-A, 0.307-A, 0.938-T and 0.916-A). Sequencing result approved the weighted voting method for this genomic position. In another example, in sample S1, at position number 25 013, the weighted voting method voted for nucleobase C based on the weighted decision-making vector of (4.698-C, 2.768-G, 6.539-C, 3.123-G, 9.275-C and 3.782-G); the ranked credibility score method called nucleobase G based on the credibility score vector of (0.99993-C, 0.9994-G, 0.9988-C, 0.9988-G, 0.9995-C and 0.99995-G). Sequencing approved the ranked credibility score method for this genomic position.
Figure 3 .

Accuracy measure of two base calling methods using sequencing result as the gold standard. Both bioinformatics methods performed excellently in relation to sequencing.
Mutation detection by tiling array
Variant SARS-CoV-2 strains add a paramount danger to the ongoing SARS-CoV-2 pandemic. When the base call for a genomic position differs from the nucleobase specified in the reference genome, a point mutation is suggested. Of the eight samples, SARS-CoV-2 tiling array detected 61 mutations at 35 genomic positions. All of these 61 mutations were recovered in the sequencing results, indicating a sensitivity of 93.8% and a specificity of 100% (Figure 4A). On average, each sample showed eight mutations. SARS-CoV-2 has four proteins: spike, envelope, membrane and nucleocapsid. From an epidemiology perspective, the spike protein is the most important protein because it is the target of current vaccines. Our analysis identified two mutated positions in the coding region of the spike protein: position 23 403 was mutated in four samples, coincident with the infamous D614G mutation; position 24 694 was mutated in two samples, without changing the amino acid outcome (Figure 4B). Another protein, nucleocapsid, has increasing importance because it has been suggested as an alternative vaccine target [15]. Our analysis identified four mutations in the coding region for nucleocapsid protein, two nonsynonymous and two synonymous (Figure 4C). The first nonsynonymous mutation appeared in sample S6 at position number 28 311, changing Pro to Leu (P13L). The second nonsynonymous mutation also appeared in sample S6 but was located at position number 28 863, changing Ser to Leu (S197L).
Figure 4 .

Tiling array detected point mutations in the SARS-CoV-2 genome. (A) Point mutations at 35 genomic positions across eight samples. Summing up mutations from individual samples; sequencing results presented 65 mutations in total, of which 61 were captured by tiling array. S1–S8: sample IDs. (B) Two mutations were identified in spike protein, one synonymous and one nonsynonymous. (C) Four mutations were identified in nucleocapsid protein, including two nonsynonymous ones.
Discussion
The SARS-CoV-2 pandemic has nearly collapsed the health care system and caused devastation to the global economy. While hope is in sight with multiple vaccines successfully completing phase III trials and are being distributed to the general public, SARS-CoV-2 variants with mutations in the spike protein ring alarm bells for complication of this pandemic. For RNA viruses, mutations generally weaken the virus, but certain mutations can increase viral transmissibility. The most infamous adverse mutation is D614G in the spike protein, which appeared first in eastern China and spread around the world soon after. This mutation has been shown to increase virion spike density and infectivity [16, 17]. Mutations like D614G cause immense concerns because they occur in the spike protein, which are the targets of current SARS-CoV-2 vaccines. Currently, all current major vaccines target the spike protein to prevent SARS-CoV-2 from entering cells. Mutations change the composition of the target protein; thus, potentially they can decrease the efficacy of SARS-CoV-2 vaccines.
Hybridization-based microarray technology was the driving force for high-throughput gene expression profiling for more than a decade. Even with the advent of high-throughput sequencing, microarray technology did not phase out but rather shifted from gene expression to genotyping [10]. With low cost and minimal maintenance, genotyping arrays are an attractive alternative to high-throughput sequencing in the identification of single-nucleotide polymorphisms and other related applications [10]. The genome tiling array is a special type of genotyping array, which aims to resequence or genotype the entire genome. Previous studies have conducted resequencing of virus genome with tiling array [18–22]. In this work, we reported the very first tiling array to resequence SARS-CoV-2 samples and detect possible mutations. To demonstrate the whole-genome genotyping accuracy, we tested eight SARS-CoV-2 samples and resorted to Illumina high-throughput sequencing for benchmarking. Both base calling algorithms we developed for the SARS-CoV-2 tiling array compared excellently to sequencing with greater than 99.5% accuracy values.
Traditional genotyping arrays seek to detect two alleles at a particular genomic position with the use of two-color probes (one color for each allele). Our SARS-CoV-2 tiling array utilizes a single-color design but leverages a four-probeset composition to detect all possible alleles at each genomic position. Of the eight samples, four had the D614G mutation in the spike protein. Our samples were collected around May 2020, a few months after the outbreak in the United States; thus, it may suggest that the D614G variant was abundant from the start of the US outbreak. We also identified two nonsynonymous mutations in the nucleocapsid protein. These two mutations might be populated at a lower frequency than the D614G mutation, as evidenced by only one of the eight samples. Nucleocapsid protein is advocated as an alternative vaccine target to spike protein. Mutations in this protein are thus highly relevant to future vaccine development.
One important lesson learned from the SARS-CoV-2 pandemic management in the process is that rapid response is key to curbing the spreading of the virus. Accurate and speedy detection of SARS-CoV-2 variants is a prerequisite to rapid response moving forward. Our innovative tiling array offers the most economical SARS-CoV-2 detection solution without sacrificing accuracy. Beyond the basic utility of SARS-CoV-2 detection, our tiling array and companion bioinformatics approaches can accurately detect emerging mutations, which are vital for future vaccine development and the harness of the pandemic.
Key Points
We designed a tiling array to capture the entire genome of SARS-CoV-2.
Using sequenced SARS-CoV-2 data as a benchmark, SARS-CoV-2 achieved 99.5% accuracy.
SARS-CoV-2 tiling array can be used to detect new SARS-CoV-2 variants.
Limin Jiang is a PhD candidate and visiting scholar at the University of New Mexico.
Yan Guo is an associate professor in the Department of Internal Medicine, University of New Mexico. He is also the director of Bioinformatics Shared Resources of the University of New Mexico. Comprehensive Cancer Center.
Hui Yu is a research fellow at the Department of Internal Medicine, University of New Mexico.
Kendall Hoff and Austin Ding are research scientists at Centrillion Biosciences.
Wei Zhou is the chief executive officer at Centrillion Biosciences.
Jeremy Edwards is a professor and the chair of the Department of Chemistry, University of New Mexico.
Contributor Information
Limin Jiang, University of New Mexico, Albuquerque, NM 87131, USA.
Yan Guo, Department of Internal Medicine, University of New Mexico, Albuquerque, NM 87131, USA.
Hui Yu, Department of Internal Medicine, University of New Mexico, Albuquerque, NM 87131, USA.
Kendal Hoff, Centrillion Biosciences, Albuquerque, NM 87131, USA.
Xun Ding, Centrillion Biosciences, Albuquerque, NM 87131, USA.
Wei Zhou, Centrillion Biosciences, Albuquerque, NM 87131, USA.
Jeremy Edwards, Department of Chemistry, University of New Mexico, Albuquerque, NM 87131, USA.
Data and code availability
R package associated with SARS-CoV-2 tiling array analysis is available at https://github.com/Limin-Jiang/Chip-for-SARS-CoV-2.
Author contributions
None.
Acknowledgments
This research was partially supported by the UNM Comprehensive Cancer Center Support Grant NCI (P30CA118100) and an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (P20GM103451).
References
- 1. Park M, Won J, Choi BY, et al. . Optimization of primer sets and detection protocols for SARS-CoV-2 of coronavirus disease 2019 (COVID-19) using PCR and real-time PCR. Exp Mol Med 2020;52(6):963–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Won J, Lee S, Park M, et al. . Development of a laboratory-safe and low-cost detection protocol for SARS-CoV-2 of the coronavirus disease 2019 (COVID-19) (vol 29, pg 107, 2020). Exp Neurobiol 2020;29(5):402–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vogels CBF, Brito AF, Wyllie AL, et al. . Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT–qPCR primer–probe sets. Nat Microbiol 2020;5(10):1299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dohla M, Boesecke C, Schulte B, et al. . Rapid point-of-care testing for SARS-CoV-2 in a community screening setting shows low sensitivity. Public Health 2020;182:170–2. doi: 10.1016/j.puhe.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Domingo E, Holland JJ. RNA virus mutations and fitness for survival. Annu Rev Microbiol 1997;51(1):151–78. [DOI] [PubMed] [Google Scholar]
- 6. Duffy S. Why are RNA virus mutation rates so damn high? PLoS Biol 2018;16(8):e3000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Drake JW, Holland JJ. Mutation rates among RNA viruses. Proc Natl Acad Sci USA 1999;96(24):13910–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koyama T, Platt D, Parida L. Variant analysis of SARS-CoV-2 genomes. Bull World Health Organ 2020;98(7):495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Washington NL, Gangavarapu K, Zeller M, et al. . Genomic epidemiology identifies emergence and rapid transmission of SARS-CoV-2 B.1.1.7 in the United States. medRxiv 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Samuels DC, Below JE, Ness S, et al. . Alternative applications of genotyping array data using multivariant methods. Trends Genet 2020;36(11):857–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009;25(14):1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li H, Handsaker B, Wysoker A, et al. . The sequence alignment/map format and SAMtools. Bioinformatics 2009;25(16):2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koch GG. Intraclass correlation coefficient. In: Kotz S, Johnson NL (eds). Encyclopedia of Statistical Sicences. New York: John Wiley & Sons, 1982, 213–7. [Google Scholar]
- 14. Guo Y, Ye F, Sheng QH, et al. . Three-stage quality control strategies for DNA re-sequencing data. Brief Bioinform 2014;15(6):879–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dutta NK, Mazumdar K, Gordy JT. The Nucleocapsid protein of SARS-CoV-2: a target for vaccine development. J Virol 2020;94(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang L, Jackson CB, Mou H, et al. . SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat Commun 2020;11(1):6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Daniloski Z, Jordan TX, Ilmain JK, et al. . The spike D614G mutation increases SARS-CoV-2 infection of multiple human cell types. Elife 2021;10. doi: 10.7554/eLife.65365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Assarsson E, Greenbaum JA, Sundstrom M, et al. . Kinetic analysis of a complete poxvirus transcriptome reveals an immediate-early class of genes. Proc Natl Acad Sci USA 2008;105(6):2140–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ayodeji M, Kulka M, Jackson SA, et al. . A microarray based approach for the identification of common foodborne viruses. Open Virol J 2009;3(1):7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim S, Jeong H, Kim EY, et al. . Genomic and transcriptomic landscape of Escherichia coli BL21(DE3). Nucleic Acids Res 2017;45(9):5285–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Al-Eitan LN, Alghamdi MA, Tarkhan AH, et al. . Genome-wide tiling array analysis of HPV-induced warts reveals aberrant methylation of protein-coding and non-coding regions. Genes (Basel) 2019;11(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sarengaowa, Hu W, Feng K, et al. . An in situ-synthesized gene chip for the detection of food-borne pathogens on fresh-cut cantaloupe and lettuce. Front Microbiol 2019;10:3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
R package associated with SARS-CoV-2 tiling array analysis is available at https://github.com/Limin-Jiang/Chip-for-SARS-CoV-2.