Fabry disease
•Download as PPTX, PDF•
29 likes•10,830 views

brief introduction about lysosomes,lysosomes storage disorders and finally fabry disease as an example of x-linked lysosomal disorder
































Fabry disease
- 1. x-linked lysosomal storage disorder
- 2. Lysosomes are sub cellular organelles responsible for the physiologic turnover of cell constituents. The lysosomes is commonly referred to as the cell’s recycling centers. They contain catabolic enzymes, which require a low pH environment in order to function optimally.
- 3. If one of these catabolic enzymes is defective, because of a mutation, large molecules accumulate within the cell, eventually killing it. Lysosomal storage diseases describe a heterogeneous group of rare inherited disorders characterized by the accumulation of undigested or partially digested macromolecules, which results in cellular dysfunction and clinical abnormalities.
- 4. Lysosomal storage disorders are caused by lysosomal dysfunction usually as a consequence of deficiency of a single enzyme required for the metabolism of lipids, glycoproteins (sugar containing proteins) or so-called mucopolysaccharides.
- 5. Classically, lysosomal storage diseases encompassed only enzyme deficiencies of the lysosomal hydrolases. More recently, the concept of lysosomal storage disease has been expanded
- 6. Recent concept : include deficiencies or defects in proteins necessary for the normal post-translational modification of lysosomal enzymes (which themselves are often glycoproteins), activator proteins, or proteins important for proper intracellular trafficking between the lysosome and other intracellular compartments.
- 9. Lysosomal storage diseases are generally classified by the accumulated substrate and include the sphingolipidoses, oligosaccharidoses, mucolipidoses, mucopolysaccharidoses (MPSs), lipoprotein storage disorders, lysosomal transport defects, neuronal ceroid lipofuscinosis and others. Age of onset and clinical manifestations may vary widely among patients with a given lysosomal storage disease.
- 10. Neuronal ceroid lipofuscinoses (NCL) is the general name for a family of at least eight genetically separate neurodegenerative disorders
- 11. Lipids are fat-like substances that are important parts of the membranes found within and between each cell and in the myelin sheath that coats and protects the nerves
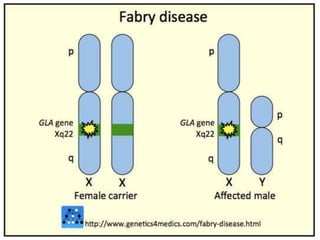
- 13. Autosomal recessive inheritance X-linked recessive inheritance occurs when the mother carries the affected gene on the X chromosome that determines the child’s gender. Affected men do not pass the disorder to their sons but their daughters will be carriers for the disorder.
- 14. Fabry disease is an inherited disorder that results from the buildup of a particular type of fat, called globotriaosylceramide http://icahn.mssm.edu/research/programs/internation al-center-for-fabry-disease/fabry-disease http://www.fabry.org/fsig.nsf/pages/fabry Fabry DiseaseSynonyms: Anderson-Fabry Disease, Alpha-Galactosidase A DeficiencyAtul Mehta, MA, MD, FRCP, FRCPath and Derralynn A Hughes, MA, DPhil, FRCP, FRCPath. in the autonomic nervous system, eyes, kidneys, and cardiovascular system. Fabry disease affects an estimated 1 in 40,000 to 60,000 males. This disorder also occurs in females, although less frequently.
- 15. Fabry disease is caused by mutations in the GLA gene. This gene provides instructions for making an enzyme called alpha-galactosidase A, which normally breaks down globotriaosylceramide GLA gene mutations that result in an absence of alpha-galactosidase A activity lead to the classic, severe form of Fabry disease. Mutations that decrease but do not eliminate the enzyme's activity usually cause the milder, late-onset forms of Fabry disease that affect only the heart or kidneys.
- 17. Diagnosis is made through clinical examination, biopsy, genetic testing, molecular analysis of cells or tissues, and enzyme assays (testing a variety of cells or body fluids for enzyme deficiency). In some forms of the disorder, a urine analysis can identify the presence of stored material. Biopsy for lipid storage disease involves removing a small sample of the liver or other tissue and studying it under a microscope
- 18. Genetic testing can help individuals who have a family history of lipid storage disease. Other genetic tests can determine if a fetus has the disorder or is a carrier of the defective gene. Prenatal testing is usually done by chorionic villus sampling, in which a very small sample of the placenta is removed and tested during early pregnancy. Results are usually available within 2 weeks.
- 19. Pain: One of the first symptoms which often begins in childhood is a painful burning sensation in the hands and feet called acroparesthesia. The pain can be severe and worsen with exercise, stress, illness, and variations in temperature.
- 20. Stomach and Intestines Early gastrointestinal symptoms of Fabry disease include abdominal cramps, frequent bowel movements shortly after eating, diarrhea, and nausea.
- 21. A common skin condition associated with Fabry disease is a red, non-painful rash known as angiokeratoma. It usually appears in the area between the belly and the knees, but may also appear on other parts of the body such as the lips, tongue, hands, and toes. Additionally, it may be confined to a small area of the body, or may affect a larger area
- 22. The surface layer of the eye (cornea) may appear abnormal when examined by an eye specialist (ophthalmologist). This unique appearance of the cornea is called cornea verticillata and while it does not affect vision, it may become more obvious with time. Cornea verticillata occurs in approximately three-quarters of patients with Fabry disease and may be a reliable indicator of the condition.
- 23. Most patients with Fabry disease will have some degree of hearing loss at some point which can either come on suddenly or develop over a period of time. Some individuals experience a ringing in the ears that is called tinnitus.
- 24. Many individuals with Fabry disease experience kidney problems, commonly beginning in adults in their mid-30s. Abnormally functioning cells in the kidney weaken the kidneys' ability to filter waste from the blood to create urine. When the kidneys cannot properly filter waste, excess protein begins to appear in the urine — a condition know as proteinuria. Over time, kidney damage may progress to the point that the kidneys lose some or all ability to function, requiring dialysis or transplant.
- 25. The heart abnormalities often described with Fabry disease include changes in the size of the heart (left ventricular enlargement), irregular heartbeat, and leaky heart valves. Such problems increase the risk of further heart complications.
- 27. troke like symptoms called transient ischemic attacks (TIAs) and in some cases, actual strokes
- 28. Pain management Daily prophylactic doses of neuropathic pain agents (eg, phenytoin, carbamazepine, gabapentin, or a combination of these agents) provide some degree of relief. They are effective in decreasing the frequency and severity of pain episodes or pain crises in most patients.Some patients may require more potent analgesics (eg, opioids) for pain management.
- 29. No specific treatment has been found to control GI symptoms in Fabry disease. However, pancrelipase, metoclopramide, H2 blockers, loperamide, and hydrochloride can ameliorate GI symptoms in some patients.Patients with abdominal symptoms often benefit from a change in eating habits that includes frequent small meals.
- 30. The results of various laser methods used to treat angiokeratomas in patients with Fabry disease have not been promising for patients who are not receiving enzyme replacement therapy (ERT). Lesions may be treated with a series of liquid nitrogen treatments prior to laser therapy.
- 31. Ocular symptoms in patients with Fabry disease rarely, if ever, cause significant impairment of vision and, as a rule, do not require treatment Hearing loss can be treated with hearing aids. Patients should avoid excessive noise exposure.
- 33. Thank you