
Preprint
Article
Exploring Silybin B as a FOXM1 Inhibitor in Glioblastoma Stem Cells: Molecular Docking, ADMET, and Molecular Dynamics Approaches
Altmetrics
Downloads
235
Views
141
Comments
0


† Equal Contribution First Authors.
This version is not peer-reviewed
Submitted:
28 June 2023
Posted:
28 June 2023
You are already at the latest version
Alerts
Abstract
Glioblastoma multiforme (GBM) is a highly heterogeneous brain tumor with limited treatment options and a poor prognosis. Cancer stem cells (CSCs) have emerged as a critical factor in GBM resistance and management, contributing to tumor growth, heterogeneity, and immunosuppression. The transcription factor FOXM1 has been identified as a key player in the progression, spread, and therapy resistance of various cancers, including GBM. In this study, researchers conducted structure-based in silico screening to identify natural compounds that could target the DNA-binding domain (DBD) of the FOXM1 protein. Through molecular docking analyses, identified Silybin B as a potential inhibitor of FOXM1, exhibiting strong interaction with the protein. MD simulations were performed to validate the binding stability of the FOXM1-Silybin B complex. The study provides valuable insights into the potential of Silybin B as a FOXM1 inhibitor and its ability to induce senescence in GBM stem cells. These findings contribute to the development of structure-based design strategies for FOXM1 inhibitors and innovative therapeutic approaches for the treatment of Glioblastoma.
Keywords:
Subject: Medicine and Pharmacology - Oncology and Oncogenics
Introduction
Glioblastoma multiforme (GBM), the most prevalent primary malignant brain tumour in people, has an estimated overall survival of less than two years. It is still an incurable disease despite the fact that there are many treatments available including surgery, chemotherapy, and radiation, as it is made up of a highly heterogeneous assortment of neoplasms (Lee et al., 2015). Therefore, identifying new targets depends on elucidating the intrinsic molecular processes underlying the abnormal features of GBM. Overexpression of Forkhead box M1 (FOXM1) promotes oncogenesis and increases invasion, angiogenesis, epithelial-mesenchymal transition, self-renewal, and chemotherapy tolerance (Halasi and Gartel, 2013; Meng et al., 2015). Due in part to the pleotropic nature of the FoxM1 pathway, the roles of FoxM1 in GBM remain poorly known.
FOXM1, a transcription factor belonging to the Forkhead box (FOX) family, is well-known for playing a crucial part in many metabolic processes, such as cell cycle control, DNA damage repair, and apoptosis (Su et al 2021). There are three distinct isoforms of the FOXM1 protein. The transcriptional activity of the FOXM1b and FOXM1c splice forms is higher than that of the FOXM1a splice variant. Through homologous recombination, the active forms of FOXM1, especially the FOXM1b and FOXM1c isoforms, can accelerate DNA double-strand repair (Myatt et al., 2014; Monteiro et al., 2013). There are several cancers, including non-small cell lung cancer, breast cancer, basal cell carcinoma, hepatocellular carcinoma, pancreatic cancer, prostate cancer, colon cancer, medulloblastoma, and GBM that are found to have increased FOXM1 expression. In GBM rodent models, high levels of FOXM1 expression in glioma cells increase tumorigenicity, invasiveness, and angiogenesis (Shimet et al., 2022). It is also reported that FOXM1found to be controlled the Wnt/β-catenin signaling pathway by stimulating the nuclear translocation of β-catenin (Zhang et al., 2011). On the other hand, it has been discovered that suppressing FOXM1 expression and activity significantly lowers the rate of cancer metastasis spread by decreasing the expression of matrix metalloproteinases 2 (MMP-2) is regarded as a crucial enzyme in the breakdown of the extracellular matrix (Dai et al., 2007). Targeting FOXM1 in Glioblastoma stem cells may prove to be a novel approach to cancer therapy and also enhance the efficacy of pharmacological therapies in GBM patients.
The identification of natural compounds that specifically target the DNA binding domain (DBD) of FOXM1 has revealed their ability to combat tumors and transform transcription factors into viable drug targets. One such compound is Silybin B, a flavonolignan that occurs naturally and is extracted from the Silybum marianum plant, commonly referred to as Milk Thistle (Soleimani et al., 2019). From thousands of years, Silybin B has been utilized in Indian Ayurvedic medicine as an effective treatment for various diseases.
According to recent research, Silybin B is extensively used extensively as a neuroprotective, hepatoprotective, cardioprotective, antioxidant, anti-cancer, anti-diabetic, anti-viral, anti-hypertensive, immunomodulator, anti-inflammatory, and photoprotective agent by targeting various cellular and molecular pathways, such as MAPK, mTOR, -catenin, and Akt (Binienda et al., 2020). It has potent pleiotropic anti-neoplastic actions against a variety of tumour cells, including glioblastoma (Baiet et al., 2018), lung (Mateen et al., 2013), renal cell carcinoma (Li et al., 2008), bladder (Zeng et al., 2011), colon (Kumar et al., 2014) and skin (Singh et al., 2005).
The purpose of this study was to better understand the antiproliferative properties of Silybin B by examining how it affects Glioblastoma stem cells' ability to undergo senescence. It was also looked into whether Silybin B's impacts on FOXM1 inhibition and cellular senescence are related. Additionally, molecular dynamics simulations (MD) were run to acquire a better understanding of the interactions between Silybin B and FOXM1. Together, these findings may shed light on how Silybin B inhibits FOXM1's transactivation activity and aid in the development of an innovative, efficient FOXM1 molecular inhibitor for the therapy of glioblastoma stem cells. The rational structure-based design of drug candidates may also benefit from a better comprehension of the binding mechanism between Silybin B and FOXM1.
Materials and Methodology
- Ligand and Protein Retrieval
3-Dimentional structures of all 98 phytochemical compounds based on their anticancer activity and 2 control compounds were downloaded in .sdf format obtained from PubChem server (https://pubchem.ncbi.nlm.nih.gov/). For FOXM1-BD protein, PDB ID: 3G73 was downloaded which is X-ray diffracted having 2.21 Å resolutions.
- Protein and Ligand Preparation
Appropriate selection and processing of coordinates for receptors and ligands are essential factors in the successful docking process. These preparation processes require the conversion of the PDB file format to the PDBQT file format. The 3D structure of FOXM1-BD with PDB ID 3G73 was then prepared for molecular docking. Chain A, water molecules, DNA, and magnesium ions were removed from the protein molecule using PyMOL software. Further, protein was minimized using Chimaera Software's AutoDock software using an AMBER ff14SB forcefield. Final protein and ligand preparation was done by assigning missing residues, hydrogen polarities, and Gasteiger and Kollman charges to proteins and ligand structures using AutoDock tools (Lim et al., 2011; Jaghoori et al., 2016).
- Molecular Docking Study
In modern drug design, molecular docking provides valuable information about drug-receptor interactions. It is frequently used to predict the binding orientation of small molecule drug candidates to their protein targets to predict the small molecule's affinity and activity. A grid box was generated, keeping the active site in consideration. The Active site was obtained using BIOVIA Discovery Studio Visualizer 2020 (https://discover.3ds.com/discovery-studio-visualizer-download) and Autoligand module of AutoDock tools to locate the coordinates for the ligand's possible binding location in the receptor's active site pocket.
Autodock Vina docking tool was used for molecular docking that takes the atomic coordinates of the target protein and selected ligand, thus predicting the most suitable docking conformation of the two. Vina's inability to replicate the docking of more than one ligand to the same target protein at once is one of its drawbacks. This is a barrier for scientists that want to use Vina to do high-throughput virtual screening. A tool called EasyDockVina was developed to make it simple and straightforward to employ numerous ligand docking against a certain protein (Mane et al., 2022; Alshehri et al., 2020). All the 100 ligand compounds were initially docked by using EasyDock Vina. During the entire docking process, the target (protein) was kept rigid while ligands were flexible with the aim to determine the best suitable pose. The selected ligands which possess considerable binding energy and following Lipinski rule were then re-docked using AutoDock Vina. An output log file with all the binding energies between protein and different orientations of ligand was further analysed. On completion of the Docking process, the highest-ranking poses with lesser binding were selected for further protein-ligand interaction analysis.
- ADMET Study andProtein-Ligand Interaction
Understanding drug Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) is crucial for drug research and development. In addition to possessing sufficient activity against the therapeutic target, a high-quality drug candidate should display the appropriate ADMET properties at a therapeutic dosage. Drug-likeness calculation was performed to know the cytotoxicity activity of compounds for human by DruLiTo open-source software (https://niper.gov.in/pi_dev_tools/DruLiToWeb/DruLiTo_index.html) (Prasanth et al., 2021; Sharma et al., 2020). Based on specific physiochemical and structural characteristics, a ligand's drug bioavailability or drug-likeness is used to determine its pharmacological importance (Metwally & Eldaly, 2022). As a result, all ligands were assessed for their druglikeness using Lipinski's five guidelines and Blood Brain Barrier crossing ability by DruLiTo software.
Only those ligands were studied for interaction analysis which passes Lipinski's five guidelines and having better binding energy. The interaction was visualized on BIOVIA Discovery Studio Visualizer 2020 (Srivastava et al., 2021). This program helps to project 3D structure to a 2D image, thus facilitating close inspection of 2D interactions of the protein-ligand complex. Moreover, to estimate final selected molecule toxicity a freely available authenticated and tested software programs ProTox II was used (https://tox-new.charite.de/protox_II/) (Mehta et al., 2022; Rachmale et al., 2022; Rolta et al., 2022).
- Molecular Dynamics Simulation
The molecular dynamics (MD) simulations method helps follow the atomic motions of molecular systems. Therefore, it's a suitable method to evaluate all the dynamic aspects of proteins, apart from the available structural information from crystallography studies (Limon et al., 2022). Although Molecular docking is a quick method to determine the binding mode of any ligand in the protein's active site, it still possesses several inaccuracies in the outcomes. Therefore, simulation is followed by docking as simulations in the system are simulated under thermal fluctuations. This technique allows studying biological and chemical systems at the atomistic level on timescales from femtoseconds to milliseconds.
MD simulations utilising Schrödinger's Desmond module were used to investigate the binding stability of top-ranked compounds at the atomic level and to comprehend the molecular interaction. The complexes of FoxM1 protein with Bryophyllin A, Silybin B, Withaferin A, Sanguinarine, and the control drugs Troglitazone were solvated using the explicit SPC water model under orthorhombic periodic boundary conditions with a buffer area of 10 between protein atoms and box edges (Zala et al., 2023; Kumar et al., 2023; Desai et al., 2023). To neutralise the charges, sodium and chloride ions were introduced into the system builder, followed by 0.15 M NaCl salt concentrations in accordance with human physiological circumstances. The construction system was then minimised using a fixed value of the OPLS3e force field to reduce electronic conflicts between protein structures and to properly align the protein structure inside the simulation bounds. Long-range electrostatic interactions were evaluated using the smooth particle mesh Ewald technique with a tolerance of 1e-09, whereas short-range Van der Waals and Coulomb interactions were calculated with a cut-off radius of 9.0 (Sudevan et al., 2022; Kikiowo et al., 2022). The simulation was ran for 100 ns under a 'isothermal-isobaric ensemble' (NPT) at a temperature of 300 K and a pressure of 1 bar after importing the built minimised system (.cms file) into the molecular dynamics module. For isothermal-isobaric conditions, the 'Nose-Hoover chain thermostat' and 'Martyna-Tobias-Klein barostat' approaches were ensembled at 100 and 200ps, respectively. Simulation snapshots were obtained at 100ps intervals, and the resultant trajectories were assessed (Aljuhani et al., 2022).
Results and Discussion
- Molecular Properties and ADME Analysis
All selected 100 ligands were analyzed for their ADME properties using DruLito software. In Table 1 molecular properties of these phytochemical compounds are shown. Employing the Lipinski rule of 5 when screening molecules with the DruLito software, which discovered 65 compounds that were suitable as lead molecules. The compounds in Table 1 that adhere to the Lipinski rule are shown by the green rows (supplementary file).
- Binding Site Prediction
Active site prediction was done by using BIOVIA Discovery Studio Visualizer 2020 and Autoligand module of AutoDock tools. The active pocket obtained from both is shown in Figure 1. The grid box was of 40 x 40 x 40 size with 0.375 Å. The selected grid center for XYZ is 3.464, 4.203, and 13.602 respectively. Major conserved residues namely ARG 297, Asp 293, ARG 280, HIS 287, TRP 308, and Leu 259 were present in binding grid which were also reported to be present in most of the literature (Dakhili et al., 2018; Zhang at el., 2022).
Molecular Docking Analysis
Prior to docking analysis we have minimized protein containing only B chain. After minimization final energy come down to -6241.25 kJ/mol from -3684.35 kJ/mol with RMSD value of 0.12459. Further, we have selected two compounds namely FDI-6 and Troglitazone as our control compounds which are widely reported to be active against the selected protein (Dakhili et al., 2018; Zhang at el., 2022). Firstly, we have done docking using EasyDock Vina (GUI of AutoDock Vina) and found that. Docked results with binding energy for 100 ligands are shown in Table 2. Only 6 ligands which follows Lipinski’s rule were found to have binding energy less than -6.6 kcal/mol. These 6 ligands were subjected to re-docking using AutoDock Vina. However, the FDI-6 compound taken as control were having only -6.0 kcal/mol so it was not taken for re-docking using AutoDock Vina. Same active site and configuration file were used to do re-docking of selected 6 ligands with target protein. Table 3 shows docked results of selected 6 ligands done by using AutoDock Vina. It is found from the docked results that Withaferin A, Bryophyllin A, Silybin B, and Sanguinarine have better binding energy in comparison to control compound Troglitazone. These 6 ligands were studied for protein-ligand interaction analysis. Out of all the selected phytochemical compounds only Withaferin A is a compound which obeys Lipinski’s rule and have binding energy -7.0 kcal/mol. Thus, this ligand is selected for further simulation studies.
- Protein-Ligand Interaction Analysis
The poses with the highest rankings following the docking procedure were chosen for further protein-ligand interaction research. All six ligands subjected to re-docking using AutoDock Vina were studied for interaction analysis. The interaction was made apparent using BIOVIA Discovery Studio Visualizer 2020. The overall interacting residues with the nature of interaction obtained from Biovia Discovery studio are presented in Table 3.
Troglitazone is an antidiabetic drug molecule which is reported to interact with (and inhibit) the FOXM1 DNA binding domain (FOXM1-BD), causing downregulation of protein expression and cancer cell proliferation inhibition. It was taken as control drug for docking analysis. Three structurally distinct FOXM1 inhibitors—troglitazone, thiostrepton, and FDI molecules appear to exert a similar binding pattern inside the FOXM1-DNA binding region (Dakhili et al., 2018) Using computer-aided drug design, Xie et al. firstly explored the binding site between FDI-6 and FOXM1 and discovered that His287, Arg236 and Ser284 were the important amino acids in the interaction (Xie et al., 2022). Figure 2 shows the two- and three-dimensional structure interacting with protein residues present in active site.
Withaferin A is a naturally occurring steroidal lactone compound derived from the Withania somnifera plant (commonly known as Ashwagandha), which has long been used in Indian Ayurvedic medicine for an effective therapy of a wide range of diseases. By increasing the expression of Bim and Bad, Withaferin A greatly reduced the growth of GBM both in vitro and in vivo and caused the intrinsic death of GBM cells (Tang et al., 2020). Withaferin A has received a lot of attention as a viable anti-neoplastic agent because of the lactonal steroid's capacity to alter a number of oncogenic pathways (Dhami et al., 2017). Figure 3 shows the two- and three-dimensional structure interacting with protein residues present in active site.
A naturally occurring substance called Bryophyllin A is present in Kalanchoe daigremontiana, Kalanchoe pinnata, and other organisms (Hernández-Caballero et al 2022). People utilise plants from the genus Kalanchoe (Family: Crassulaceae) as decorative plants all over the world in warm regions. Some of the 200 species of Kalanchoe are renowned for their ability to treat illnesses including cancer. Herbal extracts are made from a wide range of plants, and natural treatments are widely utilised throughout the world. Several of the chemical components found in Kalanchoe species, including flavonoids, bufadienolides, fatty acids, triterpenoids, alkaloids, phenolic acids, saponins, tannins, glycosides, and kalanchosides, show significant anticancer potential (Nielsen et al., 2005; Kamboj et al., 2009). Figure 4 shows the two- and three-dimensional structure interacting with protein residues present in active site.
Sanguinarine (SAG), a naturally occurring benzophenanthridine alkaloid produced from the root of the bloodroot plant Sanguinaria canadensis Linn. havingchemo-preventive properties (Xu et al., 2022). According to several studies, SANG prevents tumour metastasis and growth by interfering with a variety of cell signalling pathways and its molecular targets, including BCL-2, MAPKs, Akt, NF-B, ROS, and microRNAs (miRNAs) (Ullah et al., 2023). Figure 5 shows the two- and three-dimensional structure interacting with protein residues present in active site.
Silybin B is extensively used extensively as a neuroprotective, hepatoprotective, cardioprotective, antioxidant, anti-cancer, anti-diabetic, anti-viral, anti-hypertensive, immunomodulator, anti-inflammatory, and photoprotective agent by targeting various cellular and molecular pathways, such as MAPK, mTOR, -catenin, and Akt (Binienda et al., 2020). Figure 6 shows the two- and three-dimensional structure interacting with protein residues present in active site.
- MD Simulation Analysis
MD simulations can be utilized to evaluate the stability of protein-ligand complexes under physiological conditions. By employing MD simulations, researchers can observe and analyse the conformational changes of proteins over time, gaining valuable insights into the movements of proteins and their impact on the stability of ligands. In this study, a 100 ns MD simulations on the protein-ligand complex structures were conducted to assess the consistency of proposed phytochemicals, namely Bryophyllin A, Silybin B, Withaferin A, and Sanguinarine, within the binding cavity of the FoxM1 protein's active site. During the MD simulations, various parameters collected from the trajectory of the simulations. These parameters included Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), and Radius of Gyration (RGyr). These metrics served as indicators for evaluating the stability of the protein-ligand complex in dynamic states.
- Root-mean-square deviation (RMSD)
Root-mean-square deviation (RMSD) is a widely used metric in MD simulations to measure the average movement or deviation of atoms between two reference frames. It provides valuable information about the stability and conformational changes within protein-ligand complexes during MD simulations, typically conducted over various nanosecond timescales. Higher RMSD values for the protein backbone indicate a greater degree of unfolding or conformational changes, suggesting decreased stability of the complex. Conversely, lower RMSD values suggest a more compact and stable conformation of the protein-ligand complex (Derya et al., 2022; Halder et al., 2022). Graphical analyzing the protein Cα RMSD values, interpret the stability of the docking complex over time (Figure 7). Patterns and trends in the RMSD plot can provide valuable insights into the dynamic behavior of the protein-ligand complex, indicating regions of stability and potential conformational changes that may impact the ligand's binding within the protein's active site.
As depicted in Figure 7, the Cα RMSD demonstrates consistent stability throughout the simulation, with values ranging between 0.80 Å and 3.66 Å. The average RMSD values for the Apo FoxM1 protein and its complexes with Bryophyllin A, Silybin B, Withaferin A, Sanguinarine, and the control drug Troglitazone are 2.18 Å, 2.28 Å, 2.79 Å, 1.88 Å, 2.93 Å, and 1.93 Å, respectively. Among the complexes, notable fluctuations are observed in the FoxM1-Silybin B complex (light green colour) and the FoxM1-Sanguinarine complex (grey colour). The FoxM1-Withaferin A complex exhibits the lowest minimum RMSD value of 0.82 Å, indicating a relatively stable structure. Additionally, it has a comparatively low average RMSD value of 1.88 Å, further supporting its status as the most stable complex among the analyzed complexes. On the contrary, the FoxM1-Sanguinarine complex demonstrates the highest maximum RMSD value of 3.73 Å, indicating significant structural fluctuations. Moreover, it possesses the highest average RMSD value of 2.93 Å among the complexes, suggesting relatively lower stability compared to the others. Based on the minimal fluctuations and modest differences in RMSD values observed in the protein Cα RMSD plot, it can be concluded that the protein-ligand complexes, except for FoxM1-Sanguinarine, demonstrated stability under dynamic conditions. The minor conformational adaptations observed suggest that the complexes were able to accommodate the ligand molecules while maintaining overall stability throughout the MD simulations.
- Root-mean-square fluctuation (RMSF)
The RMSF determine the protein's residue-by-residue alterations during the course of the simulation. The RMSF value is critical for protein characterisation since it provides information about the protein's local changes as well as the protein chain. The RMSF focuses on residue fluctuations and reveals the importance of these fluctuations in the flexibility of functionally significant residues that affect the protein. In general, low RMSF values are associated with stability, while high RMSF values indicate higher flexibility. Significant alterations were seen throughout the simulation at the N-terminal regions, which are protein-flexible regions (Alsagaby et al., 2022; Ulyiye et al., 2022). During the simulation, all ligands shows interacted with around 42 amino acids of the FoxM1 protein, including Ala277, Arg236, Arg297, Asn283, Asn288, Asn302, Asp293, Asp328, Glu298, Gly280, Gly303, His287, His292, His311, Ile276, Leu259, Leu289, Leu291, Lys260, Lys278, Lys304, Met242, Met244, Phe273, Phe295, Phe307, Pro237, Pro238, Pro279, Ser240, Ser284, Ser290, Ser300, Ser306, Thr258, Thr299, Trp281, Trp308, Tyr239, Tyr241, Val296, and Val305.
The conformational changes of essential amino acids in the FoxM1 binding cavity (lowest RMSF value) demonstrated ligands capacity to generate stable interactions with the FoxM1 protein (Figure 8). All six systems (FoxM1 Apo protein, FoxM1-Bryophyllin A, FoxM1-Silybin B, FoxM1-Withaferin A, FoxM1-Sanguinarine, FoxM1-Troglitazone) demonstrated almost a similar pattern of fluctuation across the whole structure during simulation, except in FoxM1-Withaferin A and control FoxM1-Troglitazone where moderate fluctuation is observed at Glu58-Ile60 and Gly83. With a few exceptions, a slight fluctuation was observed, favoring the amino residues' stability in a dynamic state. The average RMSF values for the FoxM1 Apo protein, FoxM1-Bryophyllin A, FoxM1-Silybin B, FoxM1-Withaferin A, FoxM1-Sanguinarine, and FoxM1-Troglitazone complexes were 1.01 Å, 1.18 Å, 0.99 Å, 1.40 Å, 1.35 Å, and 0.94 Å, respectively (Table 4). These results indicate that the studied docking complexes exhibit relatively minor conformational deviations. The lower fluctuations observed in the docking complexes suggest that the residues of FoxM1 surrounding the active region have significant interactions with Bryophyllin A, Silybin B, Withaferin A, and Sanguinarine. Overall, these findings indicate that Bryophyllin A, Silybin B, Withaferin A, and Sanguinarine have the ability to interact effectively with FoxM1, forming stable complexes with limited conformational deviations.
- The radius of Gyration (RGyr)
The RGyr parameter, determined from MD simulation trajectories, is an important measure for assessing the stability of protein-ligand complexes. It represents the protein's tertiary structure and overall size, which is important for understanding its compactness and folding (Ayipo et al., 2022). During MD simulation, a constant and consistent fluctuation of RGyr suggests a well-folded protein. The RGyr graph was generated for all complexes, and no aberrant or exceptional deviations were seen in protein complexes with ligands or free apo-protein. As shown in Figure 9, the RGyr values obtained from the simulations for all three complexes were reported to be 20.3-20.74 Å. However, we observed a slight deviation in the protein-ligand complexes, which then achieved stability, indicating the system's compactness.
Based on the average RGyr values, the FoxM1 Apo protein and its complexes with Bryophyllin A, Silybin B, Withaferin A, Sanguinarine, and Troglitazone exhibit similar sizes and shapes, with average RGyr values ranging from 20.51 Å to 20.61 Å. These values indicate that, on average, the protein-ligand complexes have a relatively compact and stable structure. The close proximity of the average RGyr values suggests that the binding of Bryophyllin A, Silybin B, Withaferin A, Sanguinarine, and Troglitazone to the FoxM1 protein does not significantly alter the overall size or shape of the protein. This indicates that the complexes maintain a similar overall structure compared to the Apo protein, even in the presence of different ligands. This stability is indicative of the protein's ability to accommodate these ligands without significant structural alterations, highlighting the potential for stable interactions between the ligands and the protein's binding site.
Conclusion
In this study, we looked at the potential of several phytochemicals and employed as inhibitors against FOXM1 protein targets. Ayurvedic or herbal therapy has been utilised for hundreds of years to treat a wide range of illnesses utilising ingredients that are naturally produced. This inspired us to investigate the world of Ayurvedic medicine in search of FOXM1 inhibitors, and we found anti-proliferative properties of phytochemicals and its impact on Glioblastoma stem cells' ability to undergo senescence.
Using computational analysis, the study employed molecular docking and molecular dynamics simulations to investigated the binding of these natural compounds with the DNA binding domain (DBD) of FOXM1. The molecular docking results identified all hundreds of natural compound as a potential inhibitor of FOXM1, with favourable binding energies followed by ADMET analysis and identified 65 compounds that adhered to the Lipinski rule of 5, making them suitable as lead molecules. Further we found that Withaferin A, Bryophyllin A, Sanguinarine and Silybin B have the best chance of being effective inhibitors against FOXM1 protein after the comparison with the docking energy of control molecule. The molecular dynamics simulations further confirmed that Silybin B havind the highest stability of the protein-ligand complex and provided insights into the conformational changes and dynamics of the complex over 100 ns time. By inhibiting FOXM1 activity, Silybin B has the potential to suppress the tumorigenicity, invasiveness, and angiogenesis of glioblastoma stem cells. Furthermore, the rational structure-based design of drug candidates can benefit from the insights gained about the binding mechanism between Silybin B and FOXM1.
Financial support and sponsorship
None
Ethical approval and consent to participate
Not applicable.
Availability of data and materials
Not applicable.
Acknowledgments
Authors would like to thank the MGCUB, Motihari (Bihar) and UPES, Dehradun to provide support for this study. We would like to thank University of Manchester, United Kingdom for providing computational resources and APC Funding.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Consent for publication
Not applicable.
References
- Aljuhani, A., Ahmed, H. E., Ihmaid, S. K., Omar, A. M., Althagfan, S. S., Alahmadi, Y. M., ... & Abulkhair, H. S. (2022). In vitro and computational investigations of novel synthetic carboxamide-linked pyridopyrrolopyrimidines with potent activity as SARS-CoV-2-M Pro inhibitors. RSC advances, 12(41), 26895-26907. 2689.
- Agwupuye, J. A., Louis, H., Gber, T. E., Ahmad, I., Agwamba, E. C., Samuel, A. B., ... & Bassey, V. M. (2022). Molecular modeling and DFT studies of diazenylphenyl derivatives as a potential HBV and HCV antiviral agents. Chemical Physics Impact, 5, 100122. [CrossRef]
- Alsagaby, S. A. , Iqbal, D., Ahmad, I., Patel, H., Mir, S. A., Madkhali, Y. A., Oyouni, A., Hawsawi, Y. M., Alhumaydhi, F. A., Alshehri, B., Alturaiki, W., Alanazi, B., Mir, M. A., & Al Abdulmonem, W. (2022). In silico investigations identified Butyl Xanalterate to competently target CK2α (CSNK2A1) for therapy of chronic lymphocytic leukemia. Scientific reports, 12(1), 17648. [CrossRef]
- Ayipo, Y. O. , Ahmad, I., Alananzeh, W., Lawal, A., Patel, H., & Mordi, M. N. (2022). Computational modelling of potential Zn-sensitive non-β-lactam inhibitors of imipenemase-1 (IMP-1). Journal of biomolecular structure & dynamics, 1–21. Advance online publication. [CrossRef]
- Bai, Z. L., Tay, V., Guo, S. Z., Ren, J., & Shu, M. G. (2018). Silibinin Induced Human Glioblastoma Cell Apoptosis Concomitant with Autophagy through Simultaneous Inhibition of mTOR and YAP. BioMed research international, 2018, 6165192. [CrossRef]
- Binienda, A. , Ziolkowska, S., & Pluciennik, E. (2020). The Anticancer Properties of Silibinin: Its Molecular Mechanism and Therapeutic Effect in Breast Cancer. Anti-cancer agents in medicinal chemistry, 20(15), 1787–1796. [CrossRef]
- Dai, B. , Kang, S. H., Gong, W., Liu, M., Aldape, K. D., Sawaya, R., & Huang, S. (2007). Aberrant FoxM1B expression increases matrix metalloproteinase-2 transcription and enhances the invasion of glioma cells. Oncogene, 26(42), 6212–6219. [CrossRef]
- Derya Osmaniyea, Iqrar Ahmad, Begüm Nurpelin Sağlık Serkan Leventa, Harun M.Patel, Yusuf Ozkay, Zafer Asım Kaplancıkl, (2022) Design, Synthesis and Molecular Docking and ADME Studies of Novel Hydrazone Derivatives for AChE Inhibitory, BBB Permeability and Antioxidant Effects, Journal of Bimolecular structure and dynamic, Print ahead. [CrossRef]
- Desai, N. C. , Jadeja, D. J., Jethawa, A. M., Ahmad, I., Patel, H., & Dave, B. P. (2023). Design and synthesis of some novel hybrid molecules based on 4-thiazolidinone bearing pyridine-pyrazole scaffolds: molecular docking and molecular dynamics simulations of its major constituent onto DNA gyrase inhibition. Molecular Diversity, 1-17.
- Dhami, J. , Chang, E., & Gambhir, S. S. (2017). Withaferin A and its potential role in glioblastoma (GBM). Journal of neuro-oncology, 131(2), 201–211. [CrossRef]
- Halasi, M., & Gartel, A. L. (2013). Targeting FOXM1 in cancer. Biochemical pharmacology, 85(5), 644–652. 5. [CrossRef]
- Halder, S. K. , Ahmad, I., Shathi, J. F., Mim, M. M., Hassan, M. R., Jewel, M. J. I., Dey, P., Islam, M. S., Patel, H., Morshed, M. R., Shakil, M. S., & Hossen, M. S. (2022). A Comprehensive Study to Unleash the Putative Inhibitors of Serotype2 of Dengue Virus: Insights from an In Silico Structure-Based Drug Discovery. Molecular biotechnology, 1–14. Advance online publication. [CrossRef]
- Hernández-Caballero, M. E. , Sierra-Ramírez, J. A., Villalobos-Valencia, R., & Seseña-Méndez, E. (2022). Potential of Kalanchoe pinnata as a Cancer Treatment Adjuvant and an Epigenetic Regulator. Molecules (Basel, Switzerland), 27(19), 6425.
- Kamboj, A. , & Saluja, A. (2009). Bryophyllum pinnatum (Lam.) Kurz.: phytochemical and pharmacological profile: a review. Pharmacognosy Reviews, 3(6), 364.
- Kikiowo, B. , Ahmad, I., Alade, A. A., T Ijatuyi, T., Iwaloye, O., & Patel, H. M. (2022). Molecular dynamics simulation and pharmacokinetics studies of ombuin and quercetin against human pancreatic α-amylase. Journal of biomolecular structure & dynamics, 1–8. Advance online publication. [CrossRef]
- Kumar, S. , Oh, J. M., Abdelgawad, M. A., Abourehab, M. A., Tengli, A. K., Singh, A. K.,... & Kim, H. (2023). Development of Isopropyl-Tailed Chalcones as a New Class of Selective MAO-B Inhibitors for the Treatment of Parkinson’s Disorder. ACS omega, 8(7), 6908-6917.
- Kumar, S. , Raina, K., Agarwal, C., & Agarwal, R. (2014). Silibinin strongly inhibits the growth kinetics of colon cancer stem cell-enriched spheroids by modulating interleukin 4/6-mediated survival signals. Oncotarget, 5(13), 4972.
- Lee, Y. , Kim, K. H., Kim, D. G., Cho, H. J., Kim, Y., Rheey, J., Shin, K., Seo, Y. J., Choi, Y. S., Lee, J. I., Lee, J., Joo, K. M., & Nam, D. H. (2015). FoxM1 Promotes Stemness and Radio-Resistance of Glioblastoma by Regulating the Master Stem Cell Regulator Sox2. PloS one, 10(10), e0137703. [CrossRef]
- Li, L. , Gao, Y., Zhang, L., Zeng, J., He, D., & Sun, Y. (2008). Silibinin inhibits cell growth and induces apoptosis by caspase activation, down-regulating survivin and blocking EGFR–ERK activation in renal cell carcinoma. Cancer letters, 272(1), 61-69.
- Mateen, S. , Raina, K., & Agarwal, R. (2013). Chemopreventive and anti-cancer efficacy of silibinin against growth and progression of lung cancer. Nutrition and cancer, 65(sup1), 3-11.
- Meng, F. D. , Wei, J. C., Qu, K., Wang, Z. X., Wu, Q. F., Tai, M. H., Liu, H. C., Zhang, R. Y., & Liu, C. (2015). FoxM1 overexpression promotes epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma. World journal of gastroenterology, 21(1), 196–213. [CrossRef]
- Monteiro, L. J. , Khongkow, P., Kongsema, M., Morris, J. R., Man, C., Weekes, D., Koo, C. Y., Gomes, A. R., Pinto, P. H., Varghese, V., Kenny, L. M., Charles Coombes, R., Freire, R., Medema, R. H., & Lam, E. W. (2013). The Forkhead Box M1 protein regulates BRIP1 expression and DNA damage repair in epirubicin treatment. Oncogene, 32(39), 4634–4645. [CrossRef]
- Myatt, S. S. , Kongsema, M., Man, C. W., Kelly, D. J., Gomes, A. R., Khongkow, P., Karunarathna, U., Zona, S., Langer, J. K., Dunsby, C. W., Coombes, R. C., French, P. M., Brosens, J. J., & Lam, E. W. (2014). SUMOylation inhibits FOXM1 activity and delays mitotic transition. Oncogene, 33(34), 4316–4329. [CrossRef]
- Nielsen, A. H., Olsen, C. E., & Møller, B. L. (2005). Flavonoids in flowers of 16 Kalanchoe blossfeldiana varieties. Phytochemistry, 66(24), 2829-283.
- Shim, J. K., Lim, S. H., Jeong, J. H., Choi, R. J., Oh, Y., Park, J., Choi, S., Hong, J., Kim, S. J., Moon, J. H., Kim, E. H., Teo, W. Y., Park, B. J., Chang, J. H., Ryu, J. H., & Kang, S. G. (2022). A lignan from Alnus japonica inhibits glioblastoma tumorspheres by suppression of FOXM1. Scientific reports, 12(1), 13990. [CrossRef]
- Singh, R. P., & Agarwal, R. (2005). Mechanisms and preclinical efficacy of silibinin in preventing skin cancer. European journal of cancer, 41(13), 1969-1979.
- Soleimani, V. , Delghandi, P. S., Moallem, S. A., & Karimi, G. (2019). Safety and toxicity of silymarin, the major constituent of milk thistle extract: An updated review. Phytotherapy research : PTR, 33(6), 1627–1638. [CrossRef]
- Su, X. , Yang, Y., Yang, Q., Pang, B., Sun, S., Wang, Y., Qiao, Q., Guo, C., Liu, H., & Pang, Q. (2021). NOX4-derived ROS-induced overexpression of FOXM1 regulates aerobic glycolysis in glioblastoma. BMC cancer, 21(1), 1181. [CrossRef]
- Sudevan, S. T. , Oh, J. M., Abdelgawad, M. A., Abourehab, M. A. S., Rangarajan, T. M., Kumar, S., Ahmad, I., Patel, H., Kim, H., & Mathew, B. (2022). Introduction of benzyloxy pharmacophore into aryl/heteroaryl chalcone motifs as a new class of monoamine oxidase B inhibitors. Scientific reports, 12(1), 22404. [CrossRef]
- Tabatabaei-Dakhili, S. A. , Aguayo-Ortiz, R., Domínguez, L., & Velázquez-Martínez, C. A. (2018). Untying the knot of transcription factor druggability: Molecular modeling study of FOXM1 inhibitors. Journal of molecular graphics & modelling, 80, 197–210. [CrossRef]
- Tang, Q. , Ren, L., Liu, J., Li, W., Zheng, X., Wang, J., & Du, G. (2020). Withaferin A triggers G2/M arrest and intrinsic apoptosis in glioblastoma cells via ATF4-ATF3-CHOP axis. Cell proliferation, 53(1), e12706. [CrossRef]
- Ullah, A. , Ullah, N., Nawaz, T., & Aziz, T. (2023). Molecular Mechanisms of Sanguinarine in Cancer Prevention and Treatment. Anti-cancer agents in medicinal chemistry, 23(7), 765–778. [CrossRef]
- Ulviye Acar Çevik, Ismail Celik, Ufuk İnce, Zahra Maryam, Iqrar Ahmad, Harun Patel, Yusuf Özkay, Zafer Asım Kaplancıkl (2022), Synthesis, Biological Evaluation, and Molecular Modeling Studies of New 1,3,4-Thiadiazole Derivatives as Potent Antimicrobial Agents, Chemistry and Biodiversity. [CrossRef]
- Xie, Z. S. , Zhou, Z. Y., Sun, L. Q., Yi, H., Xue, S. T., & Li, Z. R. (2022). Structure-based virtual screening towards the discovery of novel FOXM1 inhibitors. Future medicinal chemistry, 14(4), 207–219. [CrossRef]
- Xu, R. , Wu, J., Luo, Y., Wang, Y., Tian, J., Teng, W., Zhang, B., Fang, Z., & Li, Y. (2022). Sanguinarine Represses the Growth and Metastasis of Non-small Cell Lung Cancer by Facilitating Ferroptosis. Current pharmaceutical design, 28(9), 760–768. [CrossRef]
- Zala, Ajayrajsinh R., Dhanji P. Rajani, Iqrar Ahmad, Harun Patel, and Premlata Kumari. "Synthesis, characterization, molecular dynamic simulation, and biological assessment of cinnamates linked to imidazole/benzimidazole as a CYP51 inhibitor." Journal of Biomolecular Structure and Dynamics (2023): 1-17. [CrossRef]
- Zeng, J., Sun, Y., Wu, K., Li, L., Zhang, G., Yang, Z., ... & He, D. (2011). Chemopreventive and chemotherapeutic effects of intravesical silibinin against bladder cancer by acting on mitochondria. Molecular cancer therapeutics, 10(1), 104-116.
- Zhang, N. , Wei, P., Gong, A., Chiu, W. T., Lee, H. T., Colman, H., Huang, H., Xue, J., Liu, M., Wang, Y., Sawaya, R., Xie, K., Yung, W. K., Medema, R. H., He, X., & Huang, S. (2011). FoxM1 promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer cell, 20(4), 427–442. [CrossRef]
Figure 1.
Active site pocket obtained from BIOVIA Discovery Studio and AutoDock tool.
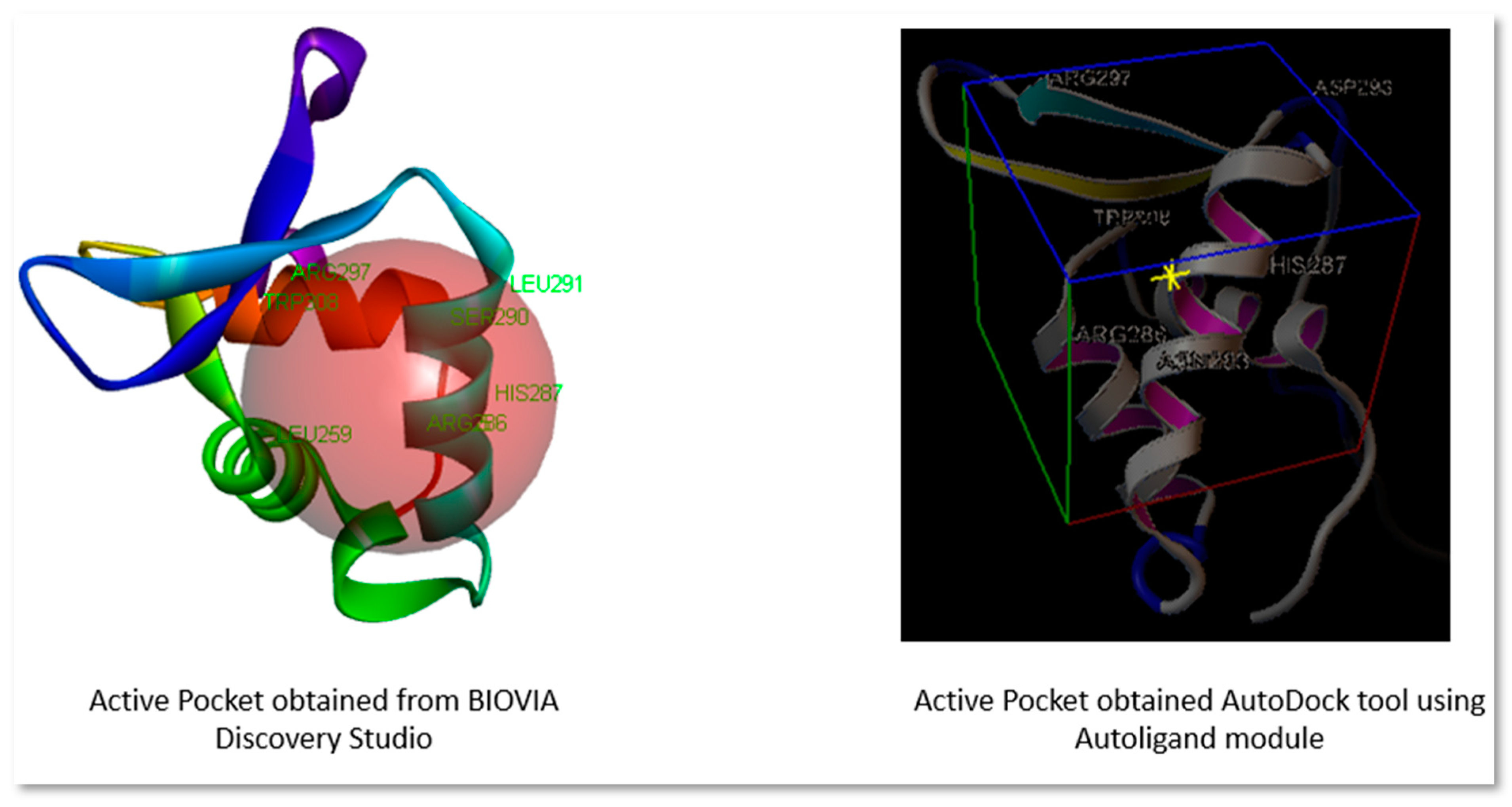
Figure 2.
Three-dimensional and Two-dimensional interaction analysis of Troglitazone with Protein. The bottom right corner of the figure displays all of the interactions, which are depicted by various colour patterns.
Figure 2.
Three-dimensional and Two-dimensional interaction analysis of Troglitazone with Protein. The bottom right corner of the figure displays all of the interactions, which are depicted by various colour patterns.
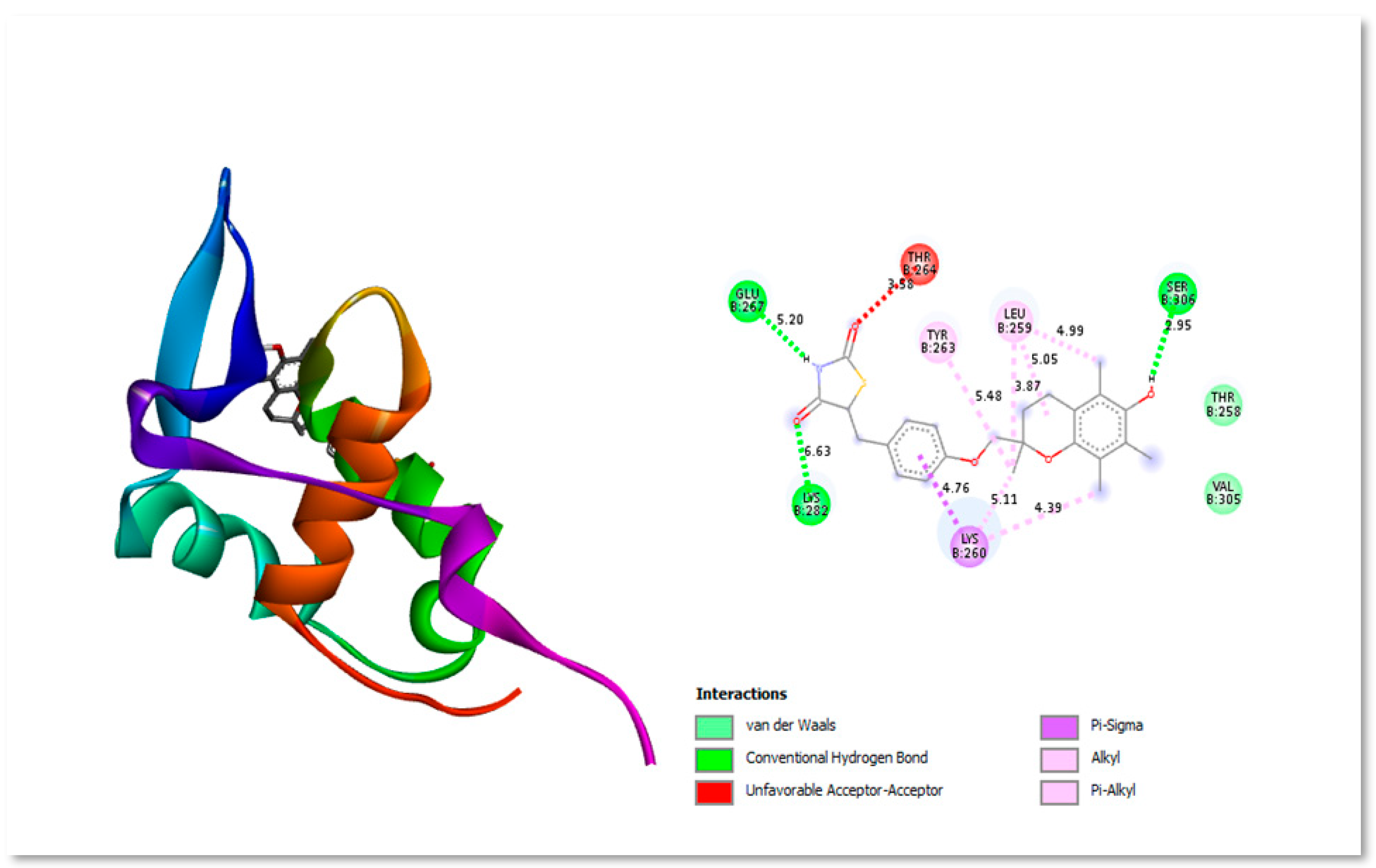
Figure 3.
Three-dimensional and Two-dimensional interaction analysis of Withaferin A with Protein. The bottom right corner of the figure displays all of the interactions, which are depicted by various colour patterns.
Figure 3.
Three-dimensional and Two-dimensional interaction analysis of Withaferin A with Protein. The bottom right corner of the figure displays all of the interactions, which are depicted by various colour patterns.

Figure 4.
Three-dimensional and Two-dimensional interaction analysis of Bryophyllin A with Protein. The bottom right corner of the figure displays all of the interactions, which are depicted by various colour patterns.
Figure 4.
Three-dimensional and Two-dimensional interaction analysis of Bryophyllin A with Protein. The bottom right corner of the figure displays all of the interactions, which are depicted by various colour patterns.

Figure 5.
Three-dimensional and Two-dimensional interaction analysis of Sanguinarine with Protein. The bottom right corner of the figure displays all of the interactions, which are depicted by various colour patterns.
Figure 5.
Three-dimensional and Two-dimensional interaction analysis of Sanguinarine with Protein. The bottom right corner of the figure displays all of the interactions, which are depicted by various colour patterns.
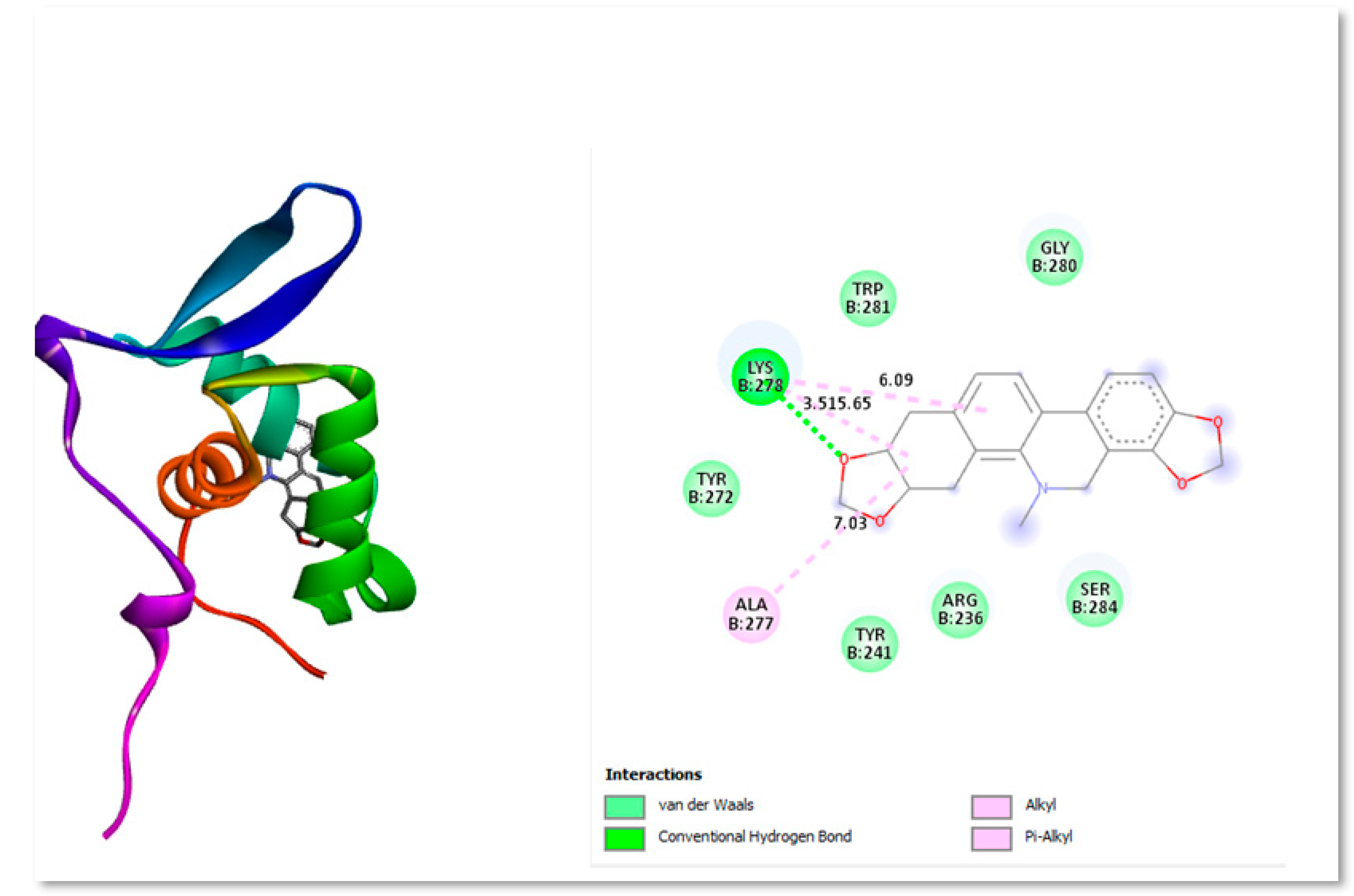
Figure 6.
Three-dimensional and Two-dimensional interaction analysis of Silybin B with Protein. The bottom right corner of the figure displays all of the interactions, which are depicted by various colour patterns.
Figure 6.
Three-dimensional and Two-dimensional interaction analysis of Silybin B with Protein. The bottom right corner of the figure displays all of the interactions, which are depicted by various colour patterns.
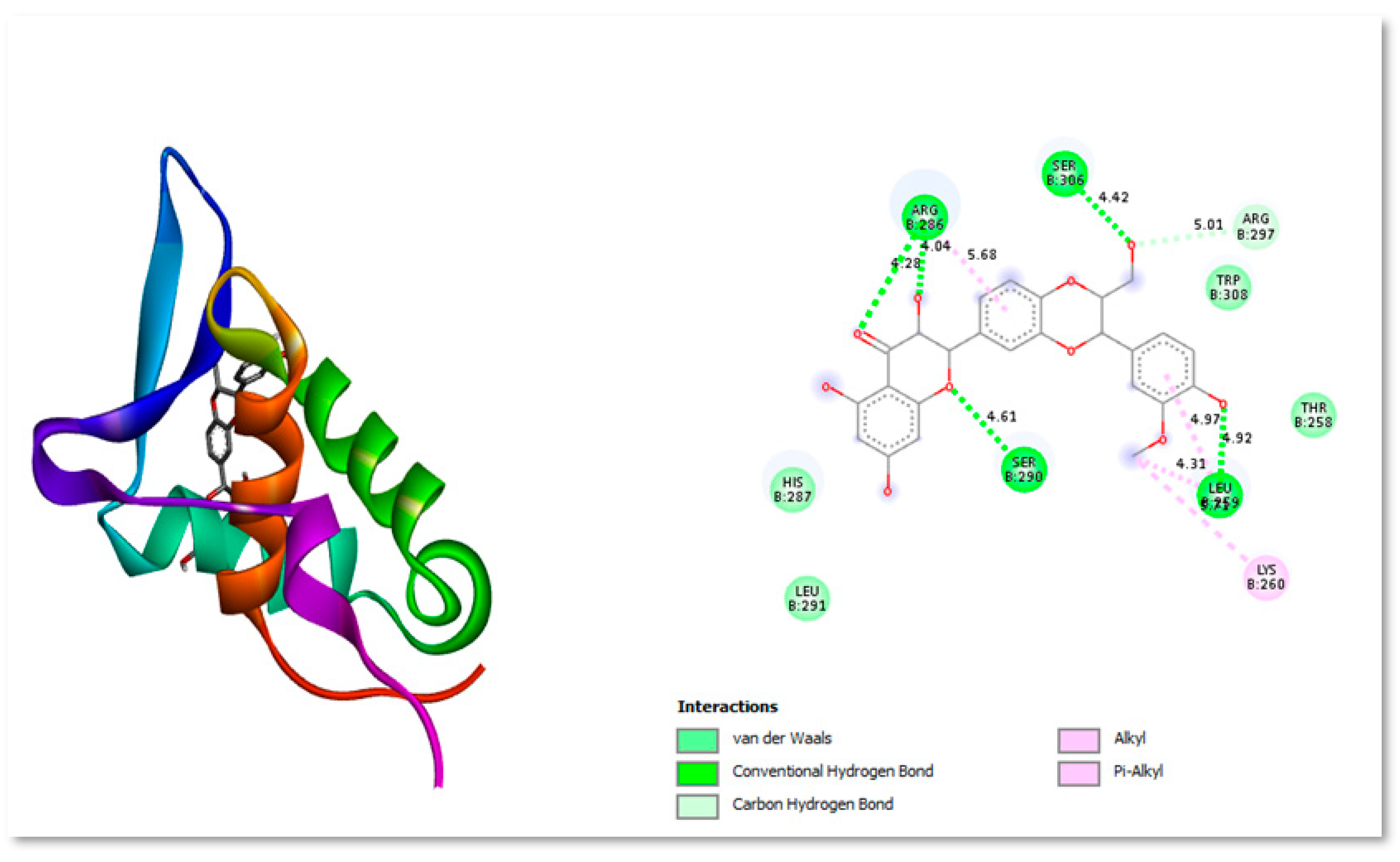
Figure 7.
Time-dependent RMSD of Cα atoms of Apo FoxM1 protein and its complexes with Bryophyllin A, Silybin B, Withaferin A, Sanguinarine, and the control drug Troglitazone.
Figure 7.
Time-dependent RMSD of Cα atoms of Apo FoxM1 protein and its complexes with Bryophyllin A, Silybin B, Withaferin A, Sanguinarine, and the control drug Troglitazone.

Figure 8.
RMSF of individual amino acids of Cα atoms of Apo FoxM1 protein and its complexes with Bryophyllin A, Silybin B, Withaferin A, Sanguinarine, and the control drug Troglitazone.
Figure 8.
RMSF of individual amino acids of Cα atoms of Apo FoxM1 protein and its complexes with Bryophyllin A, Silybin B, Withaferin A, Sanguinarine, and the control drug Troglitazone.

Figure 9.
Time-dependent Radius of Gyration (RGyr) of Apo FoxM1 protein and its complexes with Bryophyllin A, Silybin B, Withaferin A, Sanguinarine, and the control drug Troglitazone.
Figure 9.
Time-dependent Radius of Gyration (RGyr) of Apo FoxM1 protein and its complexes with Bryophyllin A, Silybin B, Withaferin A, Sanguinarine, and the control drug Troglitazone.
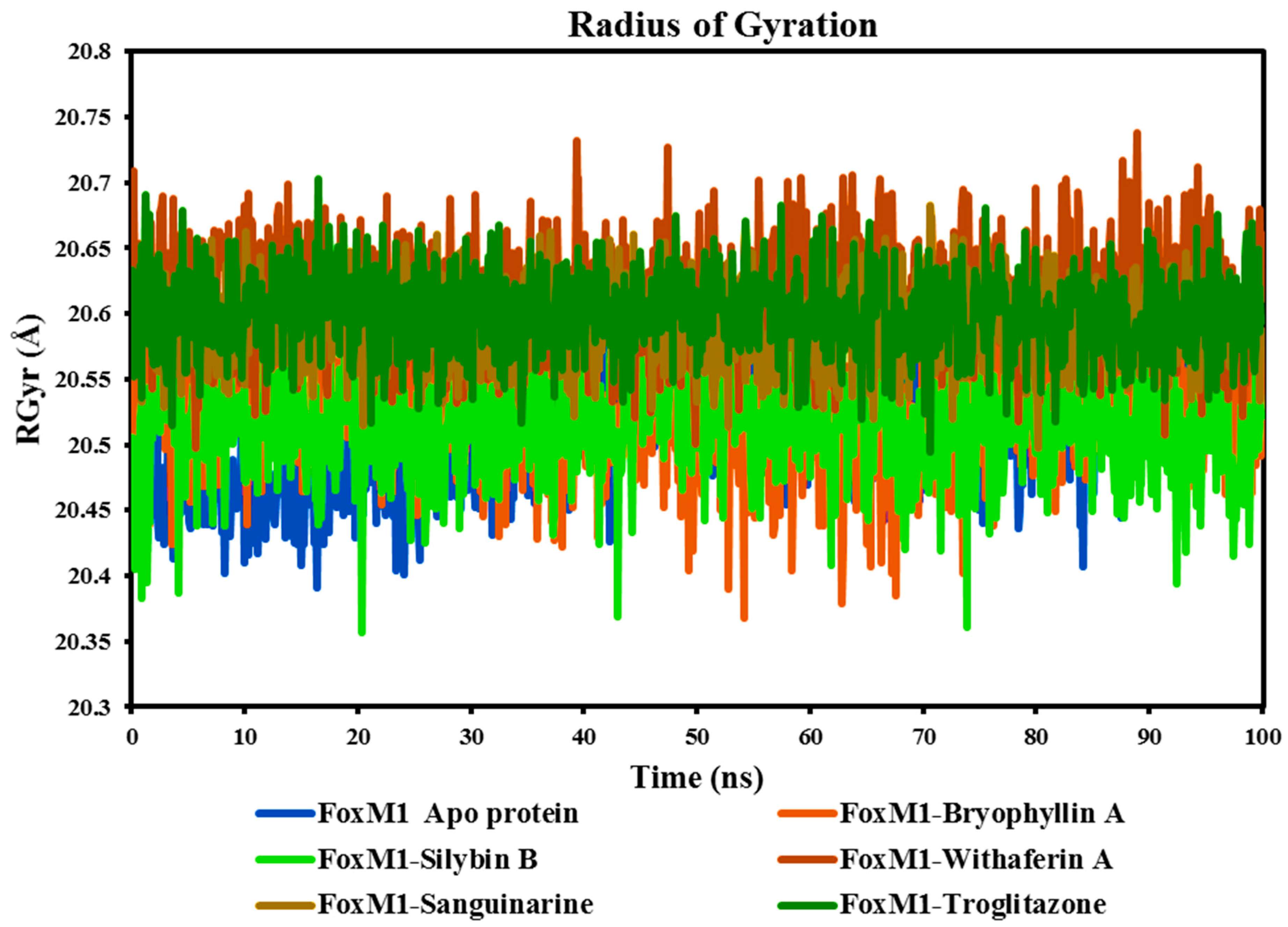

Table 2.
Docking results obtained from EasyDockVina.
| S.No. | PubChem ID | Compound name | Binding affinity |
|---|---|---|---|
| 1 | 5271805 | Ginkgetin | -7.6 |
| 2 | 10621 | Hesperidin | -7.2 |
| 3 | 5318767 | Kaempferol-3-O-beta-rutinoside | -7.1 |
| 4 | 442428 | Naringin | -7.0 |
| 5 | 65727 | Solanidine | -6.9 |
| 6 | 265237 | Withaferin A | -6.9 |
| 7 | 5488801 | Bryophyllin A | -6.9 |
| 8 | 1548994 | Silybin B | -6.8 |
| 9 | 5154 | Sanguinarine | -6.7 |
| 10 | 259846 | Lupeol | -6.7 |
| 11 | 5591 | Troglitazone (Control compound) | -6.6 |
| 12 | 24360 | Camptothecin | -6.6 |
| 13 | 64945 | Ursolic acid | -6.6 |
| 14 | 5280805 | Rutin | -6.6 |
| 15 | 5742590 | Daucosterol | -6.5 |
| 16 | 11541511 | Oleandrin | -6.5 |
| 17 | 72307 | Sesamin | -6.4 |
| 18 | 107876 | Procyanidin | -6.4 |
| 19 | 5281647 | Mangiferin | -6.4 |
| 20 | 5318645 | Isorhamnetin-3-O-glucoside | -6.4 |
| 21 | 6918260 | Larotaxel | -6.4 |
| 22 | 11250133 | Procyanidin B1 | -6.4 |
| 23 | 74978268 | Quercetin 3'-methyl ether 4'-rhamnosyl-(1->2)-glucoside | -6.4 |
| 24 | 3084131 | Paulownin | -6.3 |
| 25 | 168928 | Beta-boswellic acid | -6.2 |
| 26 | 10607 | Podofilox | -6.2 |
| 27 | 16216869 | 17-hydroxy-campesta-4,6-dien-3-one | -6.2 |
| 28 | 175267221 | Β-carotene | -6.2 |
| 29 | 3503 | Gossypol | -6.1 |
| 30 | 443654 | Peonidin-3-O-glucoside | -6.1 |
| 31 | 5281813 | Wedelolactone | -6.1 |
| 32 | 5281855 | Ellagic acid | -6.1 |
| 33 | 15895316 | Triterpene | -6.1 |
| 34 | 163184362 | 13,14-seco-stigmasta-9(11),14-dien-3alpha-ol | -6.1 |
| 35 | 65064 | Epigallocatechin-3-gallate | -6.0 |
| 36 | 92097 | Taraxerol | -6.0 |
| 37 | 5175738 | FDI 6 (Control compound) | -6.0 |
| 38 | 5280441 | Vitexin | -6.0 |
| 39 | 5280445 | Luteolin | -6.0 |
| 40 | 969516 | Curcumin | -5.9 |
| 41 | 1794427 | Chlorogenic acid | -5.9 |
| 42 | 5281316 | Cucurbitacin B | -5.9 |
| 43 | 5490064 | Avicularin | -5.9 |
| 44 | 42607963 | 8-c-Glucopyranosyleriodictylol | -5.9 |
| 45 | 932 | Naringenin | -5.8 |
| 46 | 9064 | Cianidanol | -5.8 |
| 47 | 439533 | Taxifolin | -5.8 |
| 48 | 5280343 | Quercetin | -5.8 |
| 49 | 5280443 | Apigenin | -5.8 |
| 50 | 5281605 | Baicalein | -5.8 |
| 51 | 5281672 | Myricetin | -5.8 |
| 52 | 2353 | Berberine | -5.7 |
| 53 | 23307 | Coralyne | -5.7 |
| 54 | 64981 | Arctigenin | -5.7 |
| 55 | 222284 | Beta-sitosterol | -5.7 |
| 56 | 638024 | Piperine | -5.7 |
| 57 | 5280961 | Genistein | -5.7 |
| 58 | 5319688 | 4,4′-dihydroxy-2’-methoxy-chalcone | -5.7 |
| 59 | 5281708 | Daidzein | -5.6 |
| 60 | 5281792 | Rosmarinic acid | -5.6 |
| 61 | 10807249 | Caesaldekarin J | -5.6 |
| 62 | 5316262 | Cycloolivil | -5.5 |
| 63 | 68079 | Isopimpinellin | -5.4 |
| 64 | 11414799 | Verubulin | -5.3 |
| 65 | 10205 | Plumbagin | -5.2 |
| 66 | 145858 | Anthocyanins | -5.2 |
| 67 | 442793 | Gingerol | -5 |
| 68 | 445154 | Resveratrol | -5.0 |
| 69 | 72 | Protocatechuic acid | -4.8 |
| 70 | 2214 | Apocynin | -4.8 |
| 71 | 3469 | 2,5-Dihydroxybenzoic acid | -4.8 |
| 72 | 8468 | Vanillic acid | -4.8 |
| 73 | 10742 | Syringic acid | -4.8 |
| 74 | 370 | Gallic acid | -4.7 |
| 75 | 2153 | Theophylline | -4.7 |
| 76 | 10207 | Aloe-emodin | -4.7 |
| 77 | 445858 | Ferulic acid | -4.7 |
| 78 | 637540 | O-Coumaric acid | -4.7 |
| 79 | 637775 | Sinapic acid | -4.7 |
| 80 | 689043 | Caffeic acid | -4.7 |
| 81 | 9922008 | Pipataline | -4.7 |
| 82 | 31211 | Zingerone | -4.6 |
| 83 | 637542 | P-Coumaric acid | -4.6 |
| 84 | 8655 | Syringaldehyde | -4.5 |
| 85 | 5281515 | Caryophyllene | -4.5 |
| 86 | 1183 | Vanillin | -4.4 |
| 87 | 10364 | Carvacrol | -4.4 |
| 88 | 167551 | 6-Pentadecyl salicylic acid | -4.4 |
| 89 | 444539 | Cinnamic acid | -4.4 |
| 90 | 135 | 4-Hydroxybenzoic acid | -4.3 |
| 91 | 10281 | Thymoquinone | -4.3 |
| 92 | 332 | 4-Vinylguaiacol | -4.2 |
| 93 | 938 | 3-pyridinecarboxylic acid | -4.2 |
| 94 | 126 | P-Hydroxybenzaldehyde | -3.9 |
| 95 | 289 | Catechol | -3.9 |
| 96 | 460 | Guaiacol | -3.9 |
| 97 | 68148 | Chavicol | -3.9 |
| 98 | 9793905 | S-Allyl cysteine | -3.8 |
| 99 | 5386591 | Ajoene | -3.4 |
| 100 | 65036 | Allicin | -3.1 |
Table 3.
Docking results obtained from AutoDock Vina.
| S.No. | PubChem ID | Compound name | Binding affinity |
|---|---|---|---|
| 1 | 265237 | Withaferin A | -7.0 |
| 2 | 5488801 | Bryophyllin A | -6.9 |
| 3 | 1548994 | Silybin B | -6.8 |
| 4 | 5154 | Sanguinarine | -6.7 |
| 5 | 5591 | Troglitazone (Control compound) | -6.6 |
| 6 | 24360 | Camptothecin | -6.5 |
Table 3.
Interaction study of selected ligand using Biovia Discovery Studio.
| Ligand | Interacting residues | Residues showing Vander waals’ force of attraction | Residues showing Alkyl interaction | Residues showing Carbon-Hydrogen bonding |
|---|---|---|---|---|
| Troglitazone (Control Compound) | Glu267, Lys282, Lys260, Val305, Thr 258, Tyr 263, Leu259, Ser306, and Thr264 | Val305, and Thr 258 | Thr 258, Leu259, and Lys260 | Glu267, Lys282, and Ser306 |
| Withaferin A | Ser284, Lys278, Trp281, Gly289, Ala277, Tyr272, Phe273, Tyr241, Ser240, Met242, His292, Leu291, His287, Arg236, and Asn288 | Ser284, Lys278, Trp281, Gly289, Ala277, Tyr272, Phe273, Tyr241, Ser240, Met242, His292, Leu291, and His287 | - | Arg236, and Asn288 |
| Silybin B | His287, Leu291, Thr258, Arg297, Trp308, Lys260, Arg286, Ser306, Ser290, and Leu259 | His287, Leu291, Thr258, Arg297, and Trp308 | Lys260 | Arg297, Arg286, Ser306, Ser290, and Leu259 |
| Bryophyllin A | Val305, Thr258, Lys260, Arg297, Leu289, Ser290, Ser306, Leu259, Arg286, and Trp308 | Val305, Thr258, Lys260, Arg297, Leu289, and Ser290 | Leu259, and Arg286 | Ser306 |
| Camptothecin | Leu289, Ser290, Ser306, Thr258, Asp261, Leu259, Trp308, Lys260, and Arg286 | Leu289, Ser290, Ser306, Thr258, andAsp261 | Lys260, and Arg286 | Leu259, and Trp308 |
| Sanguinarine | Lys278, Tyr272, Ala277, Tyr241, Arg236, Ser284, Gly280, and Trp281 | Tyr272, Tyr241, Arg236, Ser284, Trp281, and Gly280 | Ala277 | Lys278 |
Table 4.
The Minimum, maximum and average values of different parameters, RMSD, RMSF, RGyr, and Hydrogen Bonding of studied complexes.
Table 4.
The Minimum, maximum and average values of different parameters, RMSD, RMSF, RGyr, and Hydrogen Bonding of studied complexes.
| FoxM1 Apo protein | FoxM1-Bryophyllin A | FoxM1-Silybin B | FoxM1-Withaferin A | FoxM1-Sanguinarine | FoxM1-Troglitazone | |
|---|---|---|---|---|---|---|
| Root-mean-square deviation Å (RMSD) | ||||||
| Minimum | 0.95 | 1.03 | 0.80 | 0.82 | 0.84 | 1.11 |
| Maximum | 3.10 | 3.27 | 3.66 | 3.05 | 3.73 | 2.83 |
| Average | 2.18 | 2.28 | 2.79 | 1.88 | 2.93 | 1.93 |
| Root-mean-square fluctuation Å (RMSF) | ||||||
| Minimum | 0.46 | 0.52 | 0.47 | 0.55 | 0.59 | 0.44 |
| Maximum | 4.36 | 7.43 | 3.51 | 8.73 | 8.71 | 4.65 |
| Average | 1.01 | 1.18 | 0.99 | 1.40 | 1.35 | 0.94 |
| The radius of gyration Å (RGyr) | ||||||
| Minimum | 20.39 | 20.37 | 20.36 | 20.50 | 20.50 | 20.50 |
| Maximum | 20.63 | 20.62 | 20.62 | 20.74 | 20.68 | 20.70 |
| Average | 20.51 | 20.52 | 20.52 | 20.61 | 20.59 | 20.60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.


Exploring Silybin B as a FOXM1 Inhibitor in Glioblastoma Stem Cells: Molecular Docking, ADMET, and Molecular Dynamics Approaches
Kumari Swati
et al.
,
2023



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated