
Preprint
Article
Optimizing Chitin Extraction and Chitosan Production From Domestic Cricket Flour
Altmetrics
Downloads
184
Views
81
Comments
0
A peer-reviewed article of this preprint also exists.





This version is not peer-reviewed
Submitted:
11 January 2024
Posted:
12 January 2024
You are already at the latest version
Alerts
Abstract
Chitin and its derivative, chitosan, have diverse applications in fields such as agriculture, medicine, and biosensors, amongst others. Extraction is primarily conducted from marine sources, such as crustaceans, which have been the focus of optimization process studies. However, there are other sources that are more readily available, such as insects, where insufficient research has been conducted. The house cricket (Acheta domesticus) is a promising source for chitin extraction because of its high chitin content, availability, and short lifespan. Modern chemical chitin extraction methods have not been standardized due to the use of different reagents, molar concentrations, temperatures, and reaction times across publications. Therefore, in this study, the compositional analysis of Acheta domesticus cricket flour was determined: 2.62% humidity, 4.3% ash content, 56.29% protein, 13.35% fat, 23.44% carbohydrates, and 15.71% crude fiber content. After a drying, defatting, demineralization, deproteinization, and bleaching process, chitin extraction was performed, and chitosan was obtained via a deacetylation reaction. The demineralization process was standardized at 30ºC for 3 hours using HCl 2M, resulting in 95.85 ± 0.012%. The deproteinization process was optimized at 80ºC for 45 minutes using NaOH 2.56 M, yielding 46.37 ± 3.86%. Finally, the identity and physicochemical characteristics of the compounds were determined through characterization with FTIR, XRD, SEM and DSC.
Keywords:
Subject: Chemistry and Materials Science - Materials Science and Technology
1. Introduction
Chitin is the second most abundant polymer in nature after cellulose. This biopolymer, along with its derived compound chitosan, has diverse applications in agriculture, medicine, water treatment, cosmetics, textile, and biosensor industries, among others [1]. Due to the versatile nature of both compounds, chitin extraction and chitosan production are highly valuable processes that are generally carried out using sources such as insects, crustaceans, and fish [1,2].
The extraction of chitin is typically carried out from biological residues in the fishing industry, specifically from crustacean exoskeletons. However, these can only be obtained in sufficient quantities during seasons throughout the year, leading to limited availability and volume. Therefore, the search for an alternative source of chitin is highly relevant. Insects have been found to possess similar chitin contents to those of crustaceans, with the advantage of shorter reproductive cycles, making them an attractive new source for chitin extraction. Specifically, the cricket species Acheta domesticus has shown great promise as an alternative chitin source, as it is readily available and can be acquired in large quantities, since it is considered a pest in the agriculture industry [3].
To obtain purified chitin from crustacean, insect or fish flour, a two-step process involving demineralization (DM) and deproteinization (DP) is required. In DM step, acid hydrolysis is employed to digests the mineral content of the sample, while in DP step, the sample undergoes alkaline treatment to eliminate its proteins. Subsequently, the obtained chitin undergoes a whitening process for purification and is then transformed into chitosan through a deacetylation (DA) reaction, changing the acetamide groups into amines [3,4].
It has been shown that a wide variety of chitin extraction techniques depend on the characteristics of the source [5]. Studies consulted on chitin extraction and chitosan generation from insects reveal significant variations in reaction conditions, particularly in the concentration of reactants for both processes (ranging from 0.5 M to 1 M for DM and from 1 M to 4 M for DP). However, there is consistency in the use of sodium hydroxide and hydrochloric acid for DP and DM, respectively. The reaction time and temperature also exhibit variations, with durations ranging from 30 minutes to 2 hours for DM, 2 to 24 hours for DP, and temperatures spanning from 25 ºC to 97 ºC for DM and 82 ºC to 175 ºC for DP. These diverse reaction conditions result in chitin extraction yields ranging from 4.3% to 20% and 2.4% to 24% for chitosan obtention [6,7,8,9].
Most of the literature addressing the optimization and standardization of the chitin extraction process has focused on crustacean sources. However, there is currently no optimized and standardized chitin extraction process for insect sources, particularly for the cricket Acheta domesticus [10]. Typically, the optimization processes involve the use of statistical methods such as composite core designs, Box-Behnken response surface design, and the Taguchi method. Alternatively, new technologies like artificial neural networks (ANN) have been employed [11,12,13]. These optimization efforts have been carried out in various studies with crustacean sources. The main distinctions among them lie in the different reaction conditions, time and temperature applied to DM and DP stages of the chitin extraction process. These factors are highly specific to each crustacean species under study, and the optimization process itself depends on the statistical method employed [14,15,16,17,18,19].
Since there are currently no optimized or standardized protocols for chitin extraction and chitosan transformation for insect sources published previously and having identified the most relevant stages of the extraction process (DM and DP), the objective of this study was to establish an optimized chitin extraction protocol for insects, specifically for the cricket Acheta domesticus. Furthermore, the polymers obtained were characterized by X-Ray Diffraction (XRD), Fourier-Transform Infrared Spectroscopy (FTIR), Differential Scanning Calorimetry (DSC) and Scanning Electron Microscopy (SEM).
2. Methodology
2.1. Compositional Analysis of Cricket Flour
Cricket flour from the species Acheta domesticus was acquired from the Mexican company “Bicho”. A compositional analysis of this flour was performed (humidity, ashes, fat, protein, and crude fiber determination) according to protocols provided by the Center for Research and Protein Development (CIDPRO) from Tecnologico de Monterrey, which are based on AOAC protocols [20,21,22,23,24,25,26].
2.1.1. Humidity Determination
As the initial step in the compositional analysis, it is crucial to determine the humidity levels in the flour sample for the subsequent calculations to be reported on a dry basis. To achieve this, six samples were weighed and placed in aluminum capsules. Subsequently, they were subjected to heating in a VWRTM stove at 100ºC for 24 hours. Finally, the samples were re-weighed, and the humidity percentage was calculated using Equation 1 [20]:
2.1.2. Ash content Determination
2.1.3. Fat content Determination
The fat content determination was carried out using the Goldfish method. Five samples were weighed and placed in glass containers with 30 mL of petroleum ether in the Goldfish equipment in order to perform fat extraction for 4 hours. Once the solvent was almost completely evaporated, the containers were removed from the Goldfish extractor and placed in an oven at 100ºC overnight. Subsequently they were allowed to cool and were weighed to determine the fat content in the sample using Equation 3 [22]:
2.1.4. Protein Content Determination
The protein content was determined using the Micro Kjeldahl method, which relies on assessing the total nitrogen content. Six samples, each weighing 0.1 g of dry cricket flour, were measured alongside 2.04 to 2.05 g of digestion salts (anhydrous potassium sulphate and copper sulphate). This mixture was placed in Micro Kjeldahl flasks and allowed to react with 3.5 mL of sulphuric acid for 4 hours in pre-heated digesters. After the reaction, and when no more dark spots were visible in the flasks, the samples underwent distillation for 5 min using sodium hydroxide and a Labconco Rapid Still I distiller. Subsequently, titration was performed using 0.2N hydrochloric acid [23,24]. Finally, the percentage of protein content was determined using Equation 4 and 5, incorporating a nitrogen conversion factor of 5.25:
2.1.5. Crude fiber CONTENT Determination
Six samples underwent acid hydrolysis with 200 mL of 1.25% sulfuric acid per 1.5 g of previously degreased flour at boiling point for 30 minutes. Subsequently, the samples were filtered using dry filter papers and immersed in 200 mL of 1.25% sodium hydroxide at boiling point for an additional 30 minutes. Following this step, the samples were filtered again and rinsed with hot distilled water and 20 mL of ethanol. Next, the samples were placed on new dry ashless filter paper and placed in an oven at 60ºC for 12 hours. Afterward, they were weighed, and the crude fiber percentage was calculated by subjecting them to a furnace at 550ºC for 6 hours. The ashes were weighed, and Equation 6 was used in the calculation [25,26].
2.2. Sample Preparation
The cricket flour was treated before chitin extraction by degreasing it to eliminate interferences. This involved placing 100 g of flour with 400 mL of hexane in a VWR Rf1575 incubator at 45ºC, 125 rpm for 4 hours. This process was repeated twice. Subsequently, the sample was transferred to an aluminum tray and allowed to dry completely. To confirm the successful degreasing of the cricket flour, three samples of 2 g each were taken and analyzed using the Goldfish method to calculate the percentage of fat content using equation 3. The degreased cricket flour obtained was then used in the subsequent steps.
2.3. Chitin Extraction from Cricket Flour
The optimization and standardization process of the chitin extraction protocol was carried out in two stages: DM and DP. The purpose of each stage is to remove minerals from the degreased sample and eliminate proteins in the cricket flour, respectively. The methodology described is based on the one used by Ibitoye et al. [27].
2.3.1. Demineralization (DM)
A factorial design comprising 3 factors with 2 levels was established: temperature (30ºC and 65ºC), time (3 hours and 6 hours), and molar concentration of HCl (1M and 2M). Table 2 displays the 8 experimental conditions along with their respective results, which can be seen in the Results section.
Each experiment was performed in triplicate with 5 g samples and 50 mL of HCl. The acid concentration, temperature and time were established according to the experimental design outlined in Table 2. Samples were filtered and rinsed with hot distilled water until they reached a pH between 6 and 7. Subsequently, they were placed in an oven at 60ºC and calcinated in a furnace at 900ºC to calculate the %DM (Equation 7).
Once the battery of experiments was completed, Minitab® Version 19.2020.1.0 was used to analyse the data. An analysis of variance (ANOVA) was conducted to identify the factors that had the most significant impact on the demineralization process. Additionally, a Tukey test with a 95% confidence interval was performed to assess statistical differences among experimental conditions. This was done to select the condition that maximizes the percentage of DM with the least energy expenditure.
The degreased cricket flour was demineralized under the selected conditions. To ensure proper demineralization, three samples were taken, and the %DM was calculated using Equation 7. The subsequent steps involve the use of this demineralized flour.
2.3.2. Deproteinization (DP)
To optimize the DP process, a Box-Behnken surface response design with three factors and three levels was conducted: temperature (60 to 80ºC), time (15 to 45 min), and NaOH concentration (1M to 3M). The fifteen generated experiments are presented in Table 4.
It has been reported that the reaction time for the DP process takes hours [27]. However, due to the extreme conditions at each level for every factor, it was decided to scale down the time to minutes to preserve the integrity of the samples and to avoid wasting limited demineralized flour. The experiments were carried out in triplicate according to the conditions specified in Table 4, using 1.2 g of demineralized flour and 12 mL of NaOH. Once the reactions were completed, the samples were rinsed with hot distilled water until the pH reached a value between 7 and 8. Subsequently, they were placed in an oven at 60ºC overnight, and a protein determination assay via the Micro Kjeldahl method was performed for each sample. The percentage of deproteinization (%DP) was determined using equation 8.
Once the battery of experiments was completed, Minitab ® Version 19.2020.1.0 was used to analyze the acquired data. This analysis included processing the response surface to generate a variance analysis (ANOVA) and determining the optimized reaction condition.
The demineralized cricket flour was deproteinized using the optimized condition and dried at 60ºC in an oven overnight. The next steps involve using this deproteinized flour.
2.4. Purification and Bleaching Process
A sample of 5.65 g of deproteinized flour was placed in a flask with 56.5 mL of 1% sodium hypochlorite and left at room temperature with constant stirring for 3 hours. Subsequently, it was rinsed with distilled water and left to dry overnight in an oven at 60ºC. The same process was repeated to undergo a second bleaching process. This yielded purified chitin, from which 2.3 g were taken as samples for its physicochemical characterization.
2.5. Deacetylation Process
A sample of 1 g of purified chitin was mixed with 20 mL of 50% NaOH, the mixture was autoclaved in a Market Forge Sterilmatic ® autoclave for three cycles of 45 min at 121ºC and 0.95 kg/cm2 of pressure. Once the reaction was complete, the sample was rinsed with distilled water until it reached a neutral pH and then left to dry overnight in an oven at 60ºC. This chitosan was subsequently used for posterior physicochemical characterization. The deacetylation process was performed in triplicate.
2.6. Physicochemical Characterization Process of Chitin and Chitosan Obtained from Acheta Domesticus Cricket Flour
The obtained chitin and chitosan samples underwent analysis using Fourier-Transform Infrared Spectroscopy (FTIR), X-Ray Diffraction (XRD), Differential Scanning Calorimetry (DSC), and Scanning Electron Microscopy (SEM). For the FTIR analysis, a Perkin Elmer Spectrum 400 Spectrophotometer was employed at a resolution of 4 cm-1 and 32 scans. Equation 9 was used to estimate the degree of deacetylation (DD%) in the chitosan sample, derived from the wave number and absorbance of the carbonyl group of the secondary amide (1655 cm-1) and the hydroxyl group (3257 cm-1) [7].
The XRD analysis was done using a Rigaku MiniFlex 600 diffractometer under the following conditions: an initial angle of 5º, a final angle of 80º, 0.05º step, and a scanning velocity of 10 degrees/min. The crystallinity index (CrI %) was calculated using Equation 10, where Ic represents the maximum peak intensity for the highest peak (crystalline fraction), and Ia represents the peak with the minimum intensity within the highest peaks (amorphous fraction).
For the DSC analysis, a PerkinElmer DSC 8000 instrument was used, featuring a heating range of 25ºC to 250ºC and a heating slope of 10ºC/min. Additionally, SEM analysis was conducted using a Phenom ProX microscope operating at 5kV, equipped with a backscattered electron detector with a tungsten wire. All graphs were processed using MagicPlot ® Version 3.0.1 software.
3. Results
3.1. Compositional Analysis of Cricket Flour
Table 1 shows the consolidated results for the proximal analysis, which includes the percentages on both wet and dry bases of humidity, fat, ashes, protein, and crude fiber present in the original sample. To determine total carbohydrate content, the percentage values of humidity, fat, ashes and protein were subtracted from 100%. These values were then compared with those reported in the scientific literature for Acheta domesticus (Table 1).
3.1.1. Humidity Determination
3.1.2. Ashes Determination
Ashes determine the quantity of minerals that are present in a sample, which is important to know as a reference point to compare when the demineralization process has been carried out. A value of 4.3% was similar to that reported in the literature of 5.4% for the domestic cricket [29].
3.1.3. Fat Determination
The determined fat content in this case was 13.01% (wet basis), which is higher than the value reported by Udomsil et al. [29]. However, the differences in these values can be attributed to variations in experimental methods. Udomsil et al. [29] employed a chloroform: methanol solvent, followed by a transesterification treatment with methanolic KOH, and identified fat components using gas chromatography analysis. In contrast, this project does not involve such a detailed analysis of the starting raw material; therefore, gravimetric analysis was chosen as the preferred method.
3.1.4. Protein Determination
The protein content in cricket flour was found to be high. However, it is important to note that this value might overestimate the amount of nitrogen from amino acids due to the conversion factor for nitrogen quantity. Insects, including crickets, contain significant amounts of non-protein nitrogenated compounds such as chitin. To address this issue, we used a reference conversion factor of 5.25, as reported by Boulos et al. [30] for the species Acheta domesticus. The calculated protein value was 54.85 g, and the use of the nitrogen quantity conversion factor may account for the observed difference between our result and protein content reported by Udomsil et al. [29]. Their reported protein content was 71.7% using a conversion factor of 6.25.
3.1.5. Crude Fiber Determination
Crude fiber refers to the organic, non-nitrogenated substances that do not dissolve during subsequent acid or alkaline hydrolysis. Therefore, fiber can be reported as part of carbohydrates. In this case, cricket flour was found to have 23.44% carbohydrates, of which 67.02% is crude fiber, constituting 15.71% of the total composition of the cricket flour sample. In comparison, Udomsil et al. [29] reported values of 4.6% for fiber and 1.6% for carbohydrates. This discrepancy can be explained by the fact that these authors did not consider fiber as part of carbohydrates, and the amount of protein was likely overestimated due to the high value of the conversion factor used (6.25).
3.2. Standarization of Demineralization Process
A Tukey test was performed, revealing that experimental conditions 3, 4, 7, and 8 exhibited statistical equality and included the highest %DM values, marked in red (Table 1). Among these conditions, number seven was selected (HCl concentration 2M at 30ºC for 3 h) since it represents the shortest reaction time and the lowest reaction temperature. This choice renders the process more environmentally friendly and less energetically costly without statistically impacting the %DM.
Table 2 displays the results of DM experiments conducted to standardize this process. The percentage of demineralization (%DM) was calculated by comparing the ash content with the initial value in the proximal analysis (4.3%). A significant decrease in mineral content can be observed under all experimental conditions, with values ranging 86.79% to 93.86% for %DM.

Table 2.
DM process experimental conditions and results for Acheta domesticus cricket flour*.
| Experimental condition |
HCl concentration [M] |
Time (h) | Temperature (ºC) |
Ash content (%) | %DM |
|---|---|---|---|---|---|
| 1 | 1 | 6 | 65 | 0.37 ± 0.03 | 92.1 ± 0.69 A, B, C, D |
| 2 | 1 | 3 | 30 | 0.47 ± 0.05 | 90.09 ± 0.98 B, C, D, E |
| 3 | 2 | 3 | 65 | 0.29 ± 0.11 | 93.86 ± 2.34 A |
| 4 | 1 | 3 | 65 | 0.53 ± 0.05 | 88.84 ± 1.08 D, E |
| 5 | 1 | 6 | 30 | 0.62 ± 0.06 | 86.79 ± 1.27 E |
| 6 | 2 | 6 | 30 | 0.49 ± 0.03 | 89.56 ± 0.59 C, D, E |
| 7 | 2 | 3 | 30 | 0.36 ± 0.03 | 92.25 ± 0.67 A, B, C |
| 8 | 2 | 6 | 65 | 0.33 ± 0.04 | 93.02 ± 0.92 A, B |
*Results are the mean value of a triplicate, ± standard deviation. Mean values with different superscripts indicate statistical differences (Tukey test, 95% confidence).
Considering the obtained results, an analysis of variance (ANOVA) was conducted, revealing that the temperature and acid concentration factors were statistically significant, whereas the reaction time was not (Table 3). It was determined that a concentration of HCl at 2M and a temperature of 65ºC represent the levels that maximize the %DM, corresponding to experimental conditions 3 and 8. Condition number 8 was excluded due to observation of an unfavorable reaction in the sample (manifested as a black and viscous appearance), suggesting potential protein denaturation or alteration to the chitin structure.
Finally, the condition that maximizes the %DM was implemented to demineralize the previously degreased cricket flour. Samples were taken to perform ash determination to verify the maximization of the %DM, and the result was 95.85 ± 0.012%.
3.3. Optimization of Deproteinization Process
Table 4 displays the conditions and results of the Box-Behnken design experiments to optimize the DP process. The percentage of protein (dry weight) was used to calculate the %DP, with values ranging from 27.13 to 52.61.
Table 4.
Experimental conditions and results for deproteinization (DP) process optimization for Acheta domesticus cricket flour according to Box-Behnken design*
Table 4.
Experimental conditions and results for deproteinization (DP) process optimization for Acheta domesticus cricket flour according to Box-Behnken design*
| Experimental condition |
NaOH concentration [M] |
Time (min) | Temperature (ºC) |
Protein (%) | DP (%) |
|---|---|---|---|---|---|
| 1 | 3 | 45 | 70 | 30.67 ± 3.63 | 44.08 ± 6.44 |
| 2 | 2 | 30 | 70 | 25.99 ± 1.84 | 52.61 ± 3.27 |
| 3 | 2 | 45 | 80 | 30.3 ± 0.08 | 44.76 ± 0.14 |
| 4 | 2 | 15 | 80 | 34.15 ± 0.3 | 37.73 ± 0.53 |
| 5 | 1 | 30 | 60 | 38.57 ± 1.69 | 29.69 ± 3.00 |
| 6 | 2 | 30 | 70 | 36.69 ± 2.11 | 33.11 ± 3.75 |
| 7 | 1 | 30 | 80 | 34.64 ± 0.31 | 36.84 ± 0.54 |
| 8 | 1 | 15 | 70 | 39.97 ± 0.88 | 27.13 ± 1.56 |
| 9 | 3 | 15 | 70 | 32.27 ± 1.97 | 41.16 ± 3.50 |
| 10 | 3 | 30 | 60 | 34.39 ± 0.97 | 37.31 ± 1.73 |
| 11 | 1 | 45 | 70 | 35.32 ± 0.72 | 35.6 ± 1.27 |
| 12 | 3 | 30 | 80 | 32.13 ± 2.88 | 41.42 ± 5.11 |
| 13 | 2 | 15 | 60 | 34.22 ± 0.38 | 37.62 ± 0.68 |
| 14 | 2 | 30 | 70 | 33.54 ± 4.07 | 38.84 ± 7.22 |
| 15 | 2 | 45 | 60 | 31.43 ± 2.98 | 42.7 ± 5.30 |
* Results for protein (%) and %DP are the mean of value of a triplicate, ± standard deviation.
After conducting the 15 experiments in triplicate, a statistical analysis of the data was performed using a Box-Behnken surface response model in MINITAB® Version 19.2020.1.0 software. The resulting ANOVA used the %DP as the response variable, as depicted in Table 5. The analysis revealed that reaction time, NaOH concentration, and the concentration*concentration interaction were significant factors affecting the response variable. The regression equation for the model was determined (equation 11), where C represents the concentration of NaOH (M), T is temperature of the reaction (ºC), and t is the reaction time (minutes). Next, a prediction of the optimal conditions to maximize the %DP value was performed, resulting in the following parameters: 80ºC for temperature, 45 minutes for reaction time, and 2.56M for NaOH concentration. The %DP was predicted to reach 46.37 ± 3.86% with a 95% confidence interval, using the optimal established conditions.
The experimental value of the optimal condition was determined in triplicate to validate the %DP predicted by the model, yielding a value of 43.23 ± 1.25%. This value falls within the predicted confidence interval.
3.4. Chitin Bleaching
5.3 g of bleached purified chitin were obtained. It is noticeable that the color becomes lighter after each whitening treatment (Figure 1).
3.5. Deacetylation of Chitin and Obtention of Chitosan
After the deacetylation process of the chitin, chitosan was obtained. The degree of deacetylation was calculated using Equation 9, considering the IR bands present at 1645 cm-1 from the carbonyl group in the secondary amine and at 3237 cm-1 corresponding to the hydroxyl group of the chitosan FTIR spectrum. A deacetylation degree of 99.47% was achieved, and the presence of chitosan can be observed in the FTIR spectrum (section 4.4).
3.6. Characterization of Chitin and Chitosan Extracted from Acheta Domesticus Cricket Flour
Chitin and chitosan are valuable biopolymers for various industries. They have undergone physicochemical characterization through different techniques, including those derived from insect sources that closely resemble the cricket species Acheta domesticus. Therefore, the characterization of chitin and chitosan samples was conducted using four techniques: FTIR for identifying functional groups, and XRD, for determining the crystalline structure. Both techniques aimed to confirm the identity of the compounds. SEM, which allows determination of the morphology of the polymers, and DSC, which elucidates the thermal behavior of the sample in a range of 25 to 250ºC. The purpose of this characterization was to verify that the physicochemical properties are similar to those of commercially available chitin and chitosan.
Regarding the Fourier Transform Infrared Spectroscopy (FTIR) analysis, all bands were identified and corresponded to the characteristic functional groups of chitin and chitosan, including alcohols, amides, amines, carbonyl groups, and ethers. Also, the peaks in the X-Ray Diffraction (XRD) analysis coincided with those reported in the scientific literature (9.1º and 19.3º for chitosan at 9.1º and 19.1º in 2θ). Additionally, the Electron Scanning Microscopy (SEM) images also exhibited similar features to other analyzed samples of related cricket species. Finally, the Differential Scanning Calorimetry (DSC) analysis provided supporting evidence for the correct identification of both compounds, as indicated by the presence of exothermic peaks and the absence of endothermic peaks. These results will be further elucidated in the Discussion section.
4. Discussion
4.1. Compositional Analysis of Cricket Flour
The compositional analysis results obtained from the Acheta domesticus cricket flour differed from those reported by Udomsil et al. [29]. Regarding the ash content, the difference in values could be attributed to the sample drying time, which was 24 hours at 100ºC for the cricket flour used in our study, while Udomsil et al. [29] reported 6 hours of drying time at 105ºC. Additionally, the discrepancy may arise from the storage conditions of the samples before the dehydration process and the method implemented to determine the ash content. Our study employed a gravimetric method with furnace calcination at 900ºC in contrast to the official food method’s recommendation of 500 °C. [31]. Initially, two trials were conducted at 500ºC, but they proved usuccessful as the ashes appeared gray instead of white, and the ash percentage was too high (10.16%). Subsequent exploration of the scientific literature revealed the presence of calcium carbonate in the cricket flour, a compound challenging to fully calcinate at 500ºC, given its occurrence in insect exoskeletons. Consequently, the temperature was increased to 900ºC to ensure complete calcination [32]. On the other hand, Udomsil et al. [29] employed a different method. They used microwave digestion with hydrochloric acid and Inductively Coupled Plasma – Optical Emission Spectroscopy (ICP-OES) to quantify the minerals present in a more specific way, a technique that was beyond the scope of this study. Finally, the nutritional composition of crickets is closely linked to their maturity stage and diet [33], factors that could account for the variations in the remaining composition percentages presented in Table 1.
4.2. Standardization of Demineralization Process
Regarding the obtained results for the DM process, condition number seven was shown to be the one that maximized the %DM without compromising the environmental impact of the process (HCl concentration 2M at 30ºC for 3 h). This is supported by the analysis of variance (ANOVA) performed, whose results are shown in Table 3. Considering a significance level of 0.05, it can be observed that the statistically significant factors affecting the %DM are reaction temperature and the concentration of HCl. This is evident from their lower values in comparison to the alpha value, which were considered when determining the set of results for selection in the Tukey test.
The %DM is not commonly reported in scientific literature. However, Psarianos et al. [34] published a %DM of 91.1 ± 0.3% using 1M HCl for 2 hours at 98ºC for house crickets, in comparison to the 95.85 ± 0.012% achieved in our study. Based on these results, it can be concluded that a successful standardization of the DM process was achieved.
4.3. Optimization of Deproteinization Process
Based on the obtained results, the Box-Behnken model successfully optimized the deproteinization process to maximize the %DP. This is supported by the analysis of variance (ANOVA) results presented in Table 5. The ANOVA identified key factors influencing %DP), namely reaction temperature, time, NaOH concentration and interaction concentration*concentration. However, there was a significant low yield of solids during the deproteinization stage, obtaining 14.12% of unpurified chitin from the demineralized flour. This value cannot be directly compared to those reported in scientific literature because it does not consider the original composition of the flour. However, it is noteworthy that the yields for chitin extraction are generally low, ranging from 2.5% to 12.2% in both insects and crustaceans [35].
The %DP reflects a significant quantity of nitrogen left in the sample after deproteinization. It’s important to highlight that the Micro Kjeldahl method, employed for total nitrogen content quantification in the sample, does not exclusively measure amino acid- derived nitrogen. Chitin and chitosan, unlike proteins, are nitrogen-containing carbohydrates that exhibit fiber-like properties without being derived from vegetable sources. This characteristic might lead to an overestimation of the %DP, as it includes nitrogen from these non-protein structures, even after significant protein removal. Previously, we mentioned the possibility of using a specific nitrogen conversion factor for Acheta domesticus that accounts for these non-protein nitrogenated structures. However, in a deproteinized sample, the remaining nitrogen present is very unlikely to come from amino acids since the nitrogen conversion factor does not consider the atypically lower protein content for this species. Additionally, no comparative studies reporting %DP were found, limiting our ability to compare our findings with existing scientific literature.
Furthermore, the acid hydrolysis step in demineralization may produce hydrochlorated glucosamine (GlcHCl) in small quantities, potentially increasing the total nitrogen detected by the Micro Kjeldahl method in the cricket flour samples after deproteinization [36]. Additionally, the presence of N-acetylglucosamine, a compound that forms when chitin is exposed to highly acidic environments (like those induced by HCl), inversely affects the crystallinity index and reduces the degree of acetylation. These impacts were considered during the characterization stage as detailed in sections 4.5 and 4.7 [37,38].
In our experiments, the NaOH concentration was adequately strong, and the reaction time was sufficient across all experimental conditions to ensure efficient deproteinization. This is in line with literature reporting that the concentration of alkaline solutions for protein removal in diverse crustacean and arthropod samples typically ranges from 0.025 to 4 M of a strong base (generally NaOH). These studies have utilized temperatures ranging from 25ºC to 150ºC reaction times from 20 min to 96 h. Such conditions have been shown to yield 1.79% to 7.1% of chitin and chitosan, respectively, in cricket (Gryllus bimaculatus and Brachytrupes portentosus) samples [35].
4.4. Characterization: Identification of Chitin and Chitosan’s Functional Groups Using Fourier Transform Infrared Spectroscopy (FTIR)
FTIR analyzes how matter interacts with infrared radiation, enabling the identification of chemical species by determining the frequencies at which different functional groups absorbs in the IR spectra [39]. This technique effectively confirms the presence of both chitin and chitosan as it identifies their characteristic functional groups in the spectra. These include alcohols, amides (found in chitin), amines (in chitosan), carbonyl groups and ethers, appearing at wavelengths 3650 cm-1, 1640 cm-1, 3500 cm-1, 1700 cm-1 and ~1100 cm-1, respectively [3,40,41].
Figure 2 shows the FTIR spectrum of whitened chitin obtained under the standardized and optimized conditions demineralization and deproteinization, as outlined in previous sections. The spectrum clearly reveals the presence of all functional groups characteristic of chitin’s structure. A broad band at 3434 cm-1 indicates the stretching of the hydroxyl group (-OH), while a more intense band at 3256 cm-1, indicates the stretching of the -NH group, associated with a secondary amide, typical of chitin. Additionally, lower intensity bands at 3097 and 2876 cm-1, correspond to the stretching of the -CH and -CH3 groups, respectively. The medium intensity bands near 1645 cm-1, attributed to the carbonyl group C=O stretching, confirm the α (alpha) crystalline structure of the chitin obtained. This identification is because chitin exists in three crystalline structures in nature: α-chitin, with two bands around 1650 cm-1, β-chitin showing a single band at 1650 cm-1, and γ-chitin of which not much information can be found in the scientific literature [42].
A medium-intensity band at approximately 1551 cm-1 in the FTIR spectrum corresponds to the bending of the -NH group and the stretching of the C-N bond. Notably, the absence of a band around 1540 cm-1 in this region indicates successful deproteinization. A signal in this wave number typically indicates the presence of peptide bonds. Its absence therefore implies a minimal presence of proteins in the whitened chitin (Figure 2) [43]. The next band, at around 1375 cm-1 shows bending of the hydroxyl group bond (-OH), verifying its presence in the obtained compound as it complements the band at 3434 cm-1 (Figure 2). Finally, intense bands can be seen at 1067, 1010 and 720 cm-1 in the fingerprint region, corresponding to the stretching of the C-OH bond of the primary alcohol, the stretching of the C-O bond corresponding to the cyclic 5-carbon ether, and the bending of the N-H bond of the amide group, respectively, further delineating the molecular structure of chitin (Figure 2).
Figure 2.
IR spectrum of chitin (left) and chitosan (right) extracted from Acheta domesticus crickets showing its characteristic and dominant functional groups.
Figure 2.
IR spectrum of chitin (left) and chitosan (right) extracted from Acheta domesticus crickets showing its characteristic and dominant functional groups.
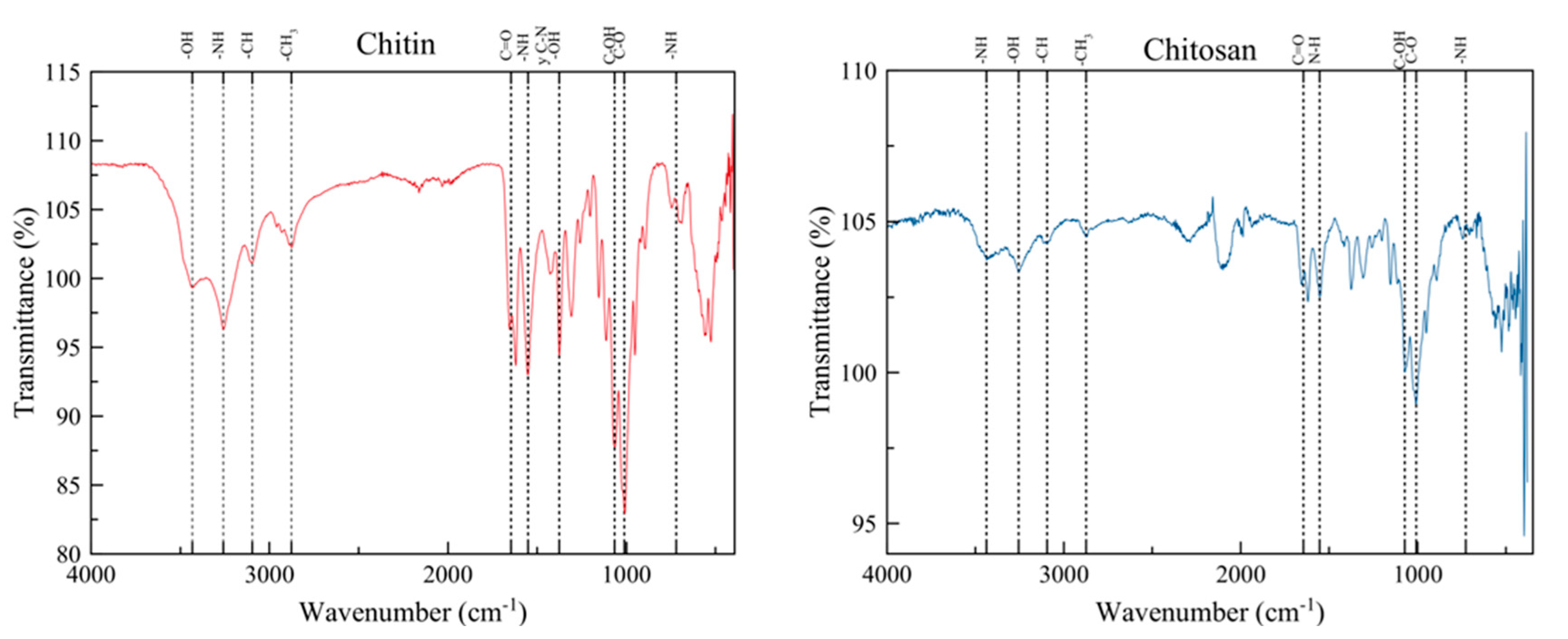
The chitosan obtained from the whitened chitin showed notable differences with the chitin IR spectrum, as shown in Figure 2 on the right side. A difference is the band at 3437 cm-1 which corresponds to the stretching of the -NH bond in the primary amine, a feature of the molecular structure of chitosan following deacetylation process of chitin. Additionally, the band at 3257 cm-1 is indicative of the hydroxyl group bond (-OH) stretching similar to that in chitin. Lower intensity bands at 3095 and 2876 cm-1, represent the stretching of the -CH and -CH3 groups, respectively. The carbonyl group stretching is observed at 1645 cm-1 and the bending of the -NH bond, characteristic of an amine, appears around 1555 cm-1 (Figure 2). These bands are typical in the IR spectra for chitosan as it is not completely deacetylated. Therefore, the presence of a carbonyl group in the IR spectrum depends on the degree of deacetylation of the chitosan the sample [8].
Since the degree of deacetylation (%DA) obtained was 99.47%, it can be considered that the time and conditions under which the deacetylation process was performed were extremely efficient. This result is comparable to those reported for chitin deacetylation from other insect sources such as silkworm Bombyx mori, in which a 94% DA was obtained using NaOH 40% for 4 h at 110ºC [35]. The results agree with reports where it is stated that the deacetylation of insects’ chitin is easier than chitin from crustacean. There are reports of only 75% DA for samples extracted from P. monodon shrimps using HCl 1M at 25ºC for demineralization, NaOH 1M at 100ºC for 8 h for deproteinization and NaOH 50% at 100ºC for 8 h for deacetylation [35].
Finally, Figure 3 shows the differences in the bands present in the chitin and chitosan that were obtained in this study (Chitin E and Chitosan E) in comparison to commercially available chitin and chitosan extracted form crab (Chitin C and Chitosan C) reported by Sáenz et al. [44].
The intensity of the stretching bands of the hydroxyl group -OH and the -NH bond around 3500 cm-1 and 3450 cm-1, respectively, are slightly more intense in the experimental chitin. However, both samples show the characteristic bands of the functional groups, with similar intensities between them, including the C=O bond stretch around 1645 cm-1, the bending of the N-H bond and the stretching of the C-N bond of the secondary amide around 1551 cm-1, the stretching of the -CH and -CH3 bonds which are visible around 3097 and 2876 cm-1, respectively, the stretching of the C-OH bond of the primary alcohol at 1067 cm-1, the stretching of the C-O group corresponding to the 5-carbon cyclic ether at 1010 cm-1 and the bending of the -NH bond in the amide group around 720 cm-1.
On the other hand, the IR spectrum for the chitosan from this work and commercial chitosan differ from each other mainly in the intensity and clarity of the characteristic bands. Nevertheless, the main functional groups can be distinguished, such as the -NH and -OH bond stretching bands at around 3400 and 3300 cm-1, respectively. The stretching of the C=O bond (1645 cm-1), the bending of the primary amine N-H bond (1555 cm-1), and the stretching of the C-O bond corresponding to the 5-carbon cyclic ether at around 1009 cm-1.
Because of the great similarity between IR spectra from chitin and chitosan from this work and commercial reference, it can be stated that we successfully obtained these polysaccharides from Acheta domesticus in accordance with the conditions established in this study [44].
Figure 3.
IR spectrum of chitin (E) and chitosan (E) extracted from Acheta domesticus cricket flour in comparison to commercially available chitin (C) and chitosan (C) reported by Sáenz et al. [44].
Figure 3.
IR spectrum of chitin (E) and chitosan (E) extracted from Acheta domesticus cricket flour in comparison to commercially available chitin (C) and chitosan (C) reported by Sáenz et al. [44].

4.5. Characterization: Identification of Chitin and Chitosan’s Crystalline Structure Using X-Ray Diffraction (XRD)
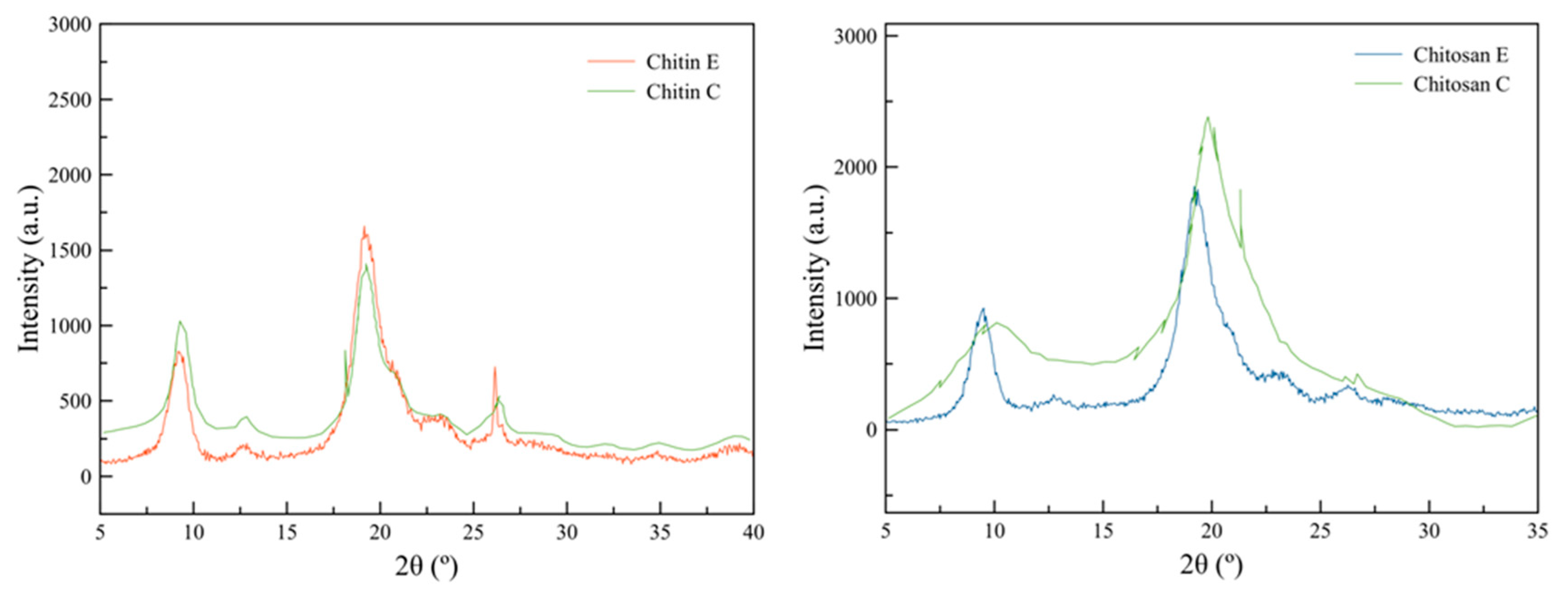
XRD is an analytical technique that measures the diffraction that an X-ray shows when it interacts with the atoms of a sample. It is especially useful to identify the crystalline structure and the purity of a sample without an extensive sample preparation [45]. In previous studies, XRD showed distinctive peaks for chitin at 9.1º and 19.3º and for chitosan at 9.1º and 19.1º in 2θ, where it is possible to identify 3 crystalline forms of chitin (α, β and γ) due to the presence of hydrogen bonds in its structure [4].
The result is a diffractogram that shows the diffraction angle of the X-ray in 2θ and the intensity of the received signal when diffracting occurs when the ray encounters the chitin and chitosan. As well, it allows the identification of the crystallographic planes and the crystalline network of both compounds [45,46].
Figure 4 shows the XRD analysis results for chitin and chitosan obtained in this study from Acheta domesticus cricket flour, compared to commercially available chitin and chitosan extracted from crab reported by Escobar et al. [14]. These diffractograms show the most representative peaks for commercial chitin and chitosan, which match, in shape and position, those found in samples obtained in this report, confirming the identity of the compounds. Additionally, the crystalline peaks around 9.6, 19.6, 21.1, and 23.7 degrees in 2θ confirm that the obtained chitin is α-chitin, as previously reported in other publications. This observation demonstrates that the crystalline form obtained aligns with the most commonly obtained form of chitin from crustaceans available in the industry [47,48].
The crystallinity index was also calculated for the samples, resulting in 93.86% and 90.60% for chitin and chitosan, respectively. In comparison, 81.84% and 58.14% were reported for commercially available chitin and chitosan. It has been reported that crystallinity indexes for chitin vary from 83.4% to 85.21% and 50.1% to 49.1% for chitosan extracted from grasshopper and shrimp husk sources, respectively [7,48]. A higher crystallinity index represents higher thermal stability for the compounds, as it indicates the presence of a minimum quantity of amorphous regions in the samples and therefore the premature degradation of its structure at high temperatures can be avoided (Section 4.7).
Figure 4.
Obtained XRD difractograms for chitin and chitosan obtained in this study (chitin E and chitosan E) from Acheta domesticus cricket flour in comparison to results reported by Escobar et al. [14] using XRD on commercially available chitin and chitosan obtained from crab (chitin C and chitosan C).
Figure 4.
Obtained XRD difractograms for chitin and chitosan obtained in this study (chitin E and chitosan E) from Acheta domesticus cricket flour in comparison to results reported by Escobar et al. [14] using XRD on commercially available chitin and chitosan obtained from crab (chitin C and chitosan C).
4.6. Morphology of Chitin and Chitosan Using Scanning Electron Microscopy (SEM)
SEM is a characterization technique that uses an electron beam instead of a beam of light to form an amplified image. It is used to perform topographical, structural, and compositional analysis. The image obtained is the response of the material to the impact of the electron beam; in other words, it is the reflection of the beam [49]. SEM images taken from previous studies show fibrous and laminar structures for chitin obtained from cricket Brachystola magna, while observing a fibrillar lattice for the subsequent chitosan [4].
Figure 5 shows the SEM obtained for the chitin extracted from Acheta domesticus cricket flour in the upper row. It is possible to identify a fibrous morphology, as well as lattice, linear and fibrous structures in its composition that confirm the crystalline structure of the compound , especially when compared to the results obtained by Monter-Miranda et al. [4]. The small and dispersed dark spots in the fibers can be attained to the degradation of the chitin. To increase the conductivity of the sample, the chitin was covered in gold, causing the impact of a larger number of electrons that magnified the image, but caused an increase in the temperature of the sample, resulting in partial degradation of the polymer.
Figure 5 also shows the SEM images obtained for the chitosan extracted from Acheta domesticus cricket flour in the lower row. Fibrillar structures similar to those present in chitin can be seen (Figure 5; upper row), which coincide with the images reported by Monter-Miranda et al. [4], and it confirms the presence of chitosan. This sample showed no dark spots, which could be because this sample was not treated with a gold coating for SEM observation in order to prevent sample degradation; however, the chitosan image was of lower detail in comparison with the chitin image (Figure 5).
4.7. Characterization: Analysis of Chitin and Chitosan Thermal Behaviour Using Differential Scanning Calorimetry (DSC)
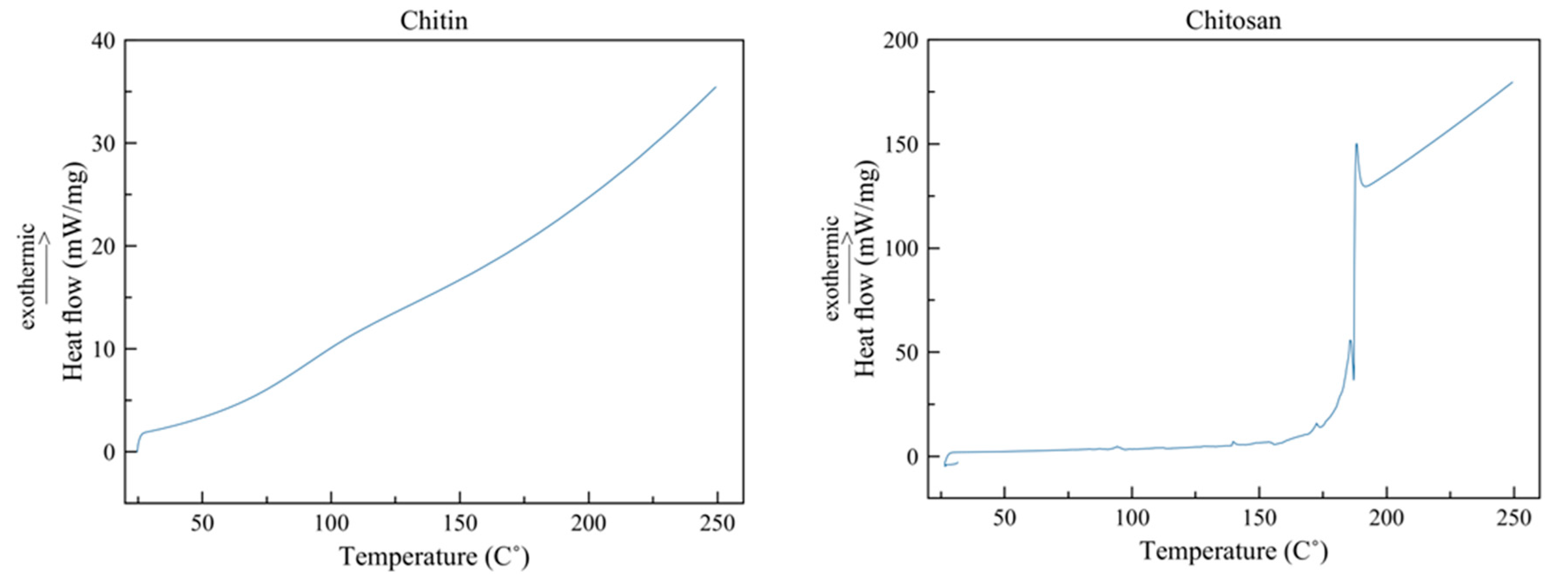
DSC was used to know the thermic stability and transition temperatures of the extracted chitin and chitosan. There were no DSC studies for these molecules from sources similar to Acheta domesticus [7,48].
Figure 6 shows the thermogram obtained by DSC of the chitin and chitosan obtained from cricket flour. In the case of chitin, no peak or slope can be perceived which may indicate the crystallization temperature or the glass transition temperature of the material. The crystallinity index of the chitin is very high and without the presence or scarce presence of amorphous regions, making the thermal behavior of the polymer very stable under high temperatures. It is estimated that changes in chitin structure may occur when surpassing 300ºC, due to decomposition of residual monomers of N-acetyl glucosamine and N-glucosamine [50]. An exothermic peak would be expected around 110 to 160ºC corresponding to water on the polymer chain [50,51]; however, since the sample was previously dried for its analysis, this could not be appreciated in the thermogram.
The chitosan thermogram shows that an exothermic peak at 185ºC corresponding to the crystallization temperature (Tc). Similar to chitin, it was expected that a significant exothermic peak may appear at 110 to 160ºC corresponding to water present in the polymer chains, this peak was visible at low intensity at around 140ºC [50,51]. The thermal behavior of chitosan is consistent with its crystalline structure. It has been reported in the scientific literature that this molecule presents a lower crystallinity index than chitin, as observed in the present study, which means that it contains more amorphous regions that turn crystalline when reaching the Tc. Therefore, chitosan is a more reactive chemical compound than chitin and consequently has a wider variety of applications (biomedicine, cosmetics, residual water treatment, etc.), being a highly sought-after compound in different industries [50,51].
Figure 6.
DSC thermogram for chitin and chitosan extracted from Acheta domesticus cricket flour.
5. Conclusions
A proximal analysis of Acheta domesticus flour revealed 2.62% moisture, 4.3% ash content, 56.29% protein, 13.35% fat, 23.44% carbohydrate and 15.71% crude fiber. An experimental protocol was proposed to enhance the efficacy of the demineralization phase and optimizing the deproteinization stage in chitin extraction from Acheta domesticus cricket flour, as well as its subsequent transformation into chitosan. The optimal conditions determined in this study were as follows: for demineralization, a temperature of 30ºC, time of 3 h, and a HCl concentration of 2M, yielding a maximum %DM of 95.85 ± 0.012%. For deproteinization, a temperature of 80ºC, 45 min reaction time, and a NaOH concentration of 2.56 M, achieving a maximum %DP of 46.37 ± 3.86%. The identity and physicochemical characteristics of the extracted chitin and produced chitosan were confirmed using FTIR, XRD, SEM and DSC analyses.
Author Contributions
Conceptualization, L.R-S, C.C-H; methodology, A.E-S, A.V-S and C.L-R; validation, L.R-S, C.C-H and L.M.M; formal analysis, A.E-S, A.V-S and C.L-R; investigation, A.E-S, A.V-S and C.L-R; resources, CC-H; data curation, L.R-S and C.C-H; writing—original draft preparation, A.E-S; writing—review and editing, L.R-S, C.C.-H and L.M.M; visualization, A.E-S; supervision, L.R-S, C.C-H and L.M.M; project administration, L.R-S and C.C-H; funding acquisition, C.C-H. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Data Availability Statement
The data presented in this study are available from the corresponding author on reasonable request.
Acknowledgments
We would like to thank M.Sc. Juan Pablo Dávila, M.Sc. Aidee Sánchez and Dr. Sergio Serna for their support in providing the necessary space, resources, and guidance for the duration of this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Knidri, H.E.; Belaabed, R.; Addaou, A.; Laajeb, A.; Lahsini, A. Extraction, chemical modification and characterization of chitin and chitosan. Int. J. Biol. Macromol. 2018, 120, 1181–1189. [Google Scholar] [CrossRef]
- Iber, B.T.; Kasan, N.A.; Torsabo, D.; Omuwa, J.W. A review of various sources of chitin and chitosan in nature. J. Renew. Mater. 2022, 10, 1097–1123. [Google Scholar] [CrossRef]
- Mohan, K.; Ganesan, A.R.; Muralisankar, T.; Jayakumar, R.; Sathishkumar, P.; Uthayakumar, V.; Revathi, N. Recent insights into the extraction, characterization, and bioactivities of chitin and chitosan from insects. Trends Food Sci. Technol. 2020, 105, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Monter-Miranda, J.G.; Tirado-Gallegos, J.M.; Zamudio-Flores, P.B.; Rios-Velasco, C.; Ornelas-Paz, J.D.J.; Salgado-Delgado, R.; Hernández-Centeno, F. Extracción y caracterización de propiedades fisicoquímicas, morfológicas y estructurales de quitina y quitosano de Brachystola magna (Girard). Rev. Mex. Ing. Quim. 2016, 15, 749–761. [Google Scholar] [CrossRef]
- Kumari, S.; Kishor, R. Chitin and chitosan: origin, properties, and applications. In Handbook of chitin and chitosan, 1st ed.; Gopi, S., Thomas, S. Pius, Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–33. [Google Scholar] [CrossRef]
- Hahn, T.; Tafi, E.; Paul, A.; Salvia, R.; Falabella, P.; Zibek, S. Current state of chitin purification and chitosan production from insects. J. Chem. Technol. Biotechnol. 2020, 95, 2775–2795. [Google Scholar] [CrossRef]
- Luo, Q.; Wang, Y.; Han, Q.; Ji, L.; Zhang, H.; Fei, Z.; Wang, Y. Comparison of the physicochemical, rheological, and morphologic properties of chitosan from four insects. Carbohydr. Polym. 2019, 209, 266–275. [Google Scholar] [CrossRef]
- Kaya, M.; Bağrıaçık, N.; Seyyar, O.; Baran, T. Comparison of chitin structures derived from three common wasp species (Vespa crabro Linnaeus, 1758, Vespa orientalis Linnaeus, 1771 and Vespula germanica (Fabricius, 1793). Arch. insect Biochem. Physiol. 2015, 89, 204–217. [Google Scholar] [CrossRef]
- Kaya, M.; Bitim, B.; Mujtaba, M.; Koyuncu, T. Surface morphology of chitin highly related with the isolated body part of butterfly (Argynnis pandora). Int. J. Biol. Macromol. 2015, 81, 443–449. [Google Scholar] [CrossRef]
- Amoo, K.O.; Olafadehan, O.A.; Ajayi, T.O. Optimization studies of chitin and chitosan production from Penaeus notialis shell waste. Afr. J. Biotechnol. 2019, 18, 670–688. [Google Scholar] [CrossRef]
- Ovando, E.; Rodríguez-Sifuentes, L.; Martínez, L.M.; Chuck-Hernández, C. Optimization of soybean protein extraction using by-products from NaCl electrolysis as an application of the industrial symbiosis concept. Appl. Sci. 2022 12, 3113. [CrossRef]
- Dar, M.A.; Kaushik, G. Optimization of process parameters for biodegradation of malathion by Micrococcus aloeverae MAGK3 using taguchi methodology and metabolic pathway analysis. Biocatal. Agric. Biotechnol. 2022, 42, 102362. [Google Scholar] [CrossRef]
- Lahiri, D.; Nag, M.; Sarkar, T.; Dutta, B.; Ray, R.R. Antibiofilm activity of α-amylase from Bacillus subtilis and prediction of the optimized conditions for biofilm removal by response surface methodology (RSM) and artificial neural network (ANN). Appl. Biochem. Biotechnol. 2022, 193, 1853–1872. [Google Scholar] [CrossRef] [PubMed]
- Sierra, D.M.E.; Orozco, C.P.O.; Rodríguez, M.A.Q.; Villa, W.A.O. Optimización de un protocolo de extracción de quitina y quitosano desde caparazones de crustáceos. Sci. Tech. 2013, 18, 260–266. [Google Scholar] [CrossRef]
- Tokatlı, K.; Demirdöven, A. Optimization of chitin and chitosan production from shrimp wastes and characterization. J. Food process. Preserv. 2018, 42, e13494. [Google Scholar] [CrossRef]
- Gîjiu, C.L.; Isopescu, R.; Dinculescu, D.; Memecică, M.; Apetroaei, M.R.; Anton, M.; Rău, I. Crabs marine waste—a valuable source of chitosan: tuning chitosan properties by chitin extraction optimization. Polymers 2022, 14, 4492. [Google Scholar] [CrossRef] [PubMed]
- Dinculescu, D.; Gîjiu, C.L.; Apetroaei, M.R.; Isopescu, R.; Rău, I.; Schröder, V. Optimization of chitosan extraction process from Rapana venosa egg capsules waste using experimental design. Materials 2023, 16, 525. [Google Scholar] [CrossRef]
- Pădurețu, C.C.; Isopescu, R.D.; Gîjiu, C.L.; Rău, I.; Apetroaei, M.R.; Schröder, V. Optimization of chitin extraction procedure from shrimp waste using Taguchi method and chitosan characterization. Mol. Crys. Liq. Crys. 2019, 695, 19–28. [Google Scholar] [CrossRef]
- Dong, F.; Qiu, H.; Jia, S.; Dai, C.; Kong, Q.; Xu, C. Optimization of extraction of chitin from procambarus clarkia shell by Box-Behnken design. 4th International Conference on Energy Materials and Environment Engineering (ICEMEE 2018). Zhuhai, China, September 28–30 2018; E3S Web of Conferences 38, 02010 (2018). Page 5. 28 September.
- AOAC Official Method 934.06. Moisture in dried fruits. Rockville, MD, USA: AOAC International. 1996.
- AOAC 923.03 Ashes. Available on line: https://law.resource.org/pub/us/cfr/ibr/002/aoac.methods.1.1990.pdf. (Accessed: Nov. 25, 2023).
- AACC. International Approved Methods of the American Association of Cereal Chemists; Minnesota University Press: St. Paul, MN, USA, 2000. [Google Scholar]
- AOAC Official Method 984.13. Protein (crude) in animal feed and pet food. Rockville, MD, USA: AOAC International. 1996.
- NOM-Y-118-A-1982 Determinación de proteína. Available on line: https://www.dof.gob.mx/nota_detalle.php?codigo=4790976&fecha=04/01/1983#gsc.tab=0 (Accessed: Nov. 28, 2023).
- NOM-F-90-S-1978 Determinación de Fibra Cruda en Alimentos. Available on line: https://www.dof.gob.mx/nota_detalle.php?codigo=4799842&fecha=27/03/1979#gsc.tab=0 (Accessed: Nov. 25, 2023).
- AOAC Official method 962.09. Crude fiber in animal feed. MD, USA: AOAC International. 2005.
- Ibitoye, E.B.; Lokman, I.H.; Hezmee, M.N.M.; Goh, Y.M.; Zuki, A.B.Z.; Jimoh, A.A. Extraction and physicochemical characterization of chitin and chitosan isolated from house cricket. Biomed. Mater. 2018, 13, 025009. [Google Scholar] [CrossRef]
- Nielsen, S.S. Food analysis laboratory manual, 3rd ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2003; p. 557. [Google Scholar]
- Udomsil, N.; Imsoonthornruksa, S.; Gosalawit, C.; Ketudat-Cairns, M. Nutritional values and functional properties of house cricket (Acheta domesticus) and field cricket (Gryllus bimaculatus). Food Sci.Technol.Res. 2019, 25, 597–605. [Google Scholar] [CrossRef]
- Boulos, S.; Tännler, A.; Nyström, L. Nitrogen-to-protein conversion factors for edible insects on the Swiss market: T. molitor, A. domesticus, and L. migratoria. Front Nutr. 2020, 7, 89. [Google Scholar] [CrossRef]
- AOAC. Official Methods of Analysis, 21st ed.; AOAC International: Gaithersburg, MD, USA, 2019. [Google Scholar]
- Hahn, T.; Tafi, E.; Paul, A.; Salvia, R.; Falabella, P.; Zibek, S. Current state of chitin purification and chitosan production from insects. J. Chem. Technol. Biotechnol. 2020, 95, 2775–2795. [Google Scholar] [CrossRef]
- Bawa, M.; Songsermpong, S.; Kaewtapee, C.; Chanput, W. Effect of diet on the growth performance, feed conversion, and nutrient content of the house cricket. J. Insect Sci. 2020, 20, 10. [Google Scholar] [CrossRef] [PubMed]
- Psarianos, M.; Ojha, S.; Schneider, R.; Schlüter, O.K. Chitin isolation and chitosan production from house crickets (Acheta domesticus) by environmentally friendly methods. Molecules 2022, 27, 5005. [Google Scholar] [CrossRef] [PubMed]
- Zainol Abidin, N.A.; Kormin, F.; Zainol Abidin, N.A.; Mohamed Anuar, N.A.F.; Abu Bakar, M.F. The potential of insects as alternative sources of chitin: An overview on the chemical method of extraction from various sources. Int. J. Mol. Sci. 2020, 21, 4978. [Google Scholar] [CrossRef]
- Moreno Villa, F.A. Obtención de glucosamina hidroclorada (GIcHCI) a partir de la hidrólisis ácida de quitina y quitosano extraidos de desecho de jaiba (Callinectes arcuatus). Master's thesis. Universidad de Sonora. Hermosillo, Sonora, México, June 30th, 2010.
- Lerma, A.G.V. Modificación estructural de quitina mediante métodos físicos y químicos para su hidrólisis enzimática mediante quitinasas de Lecanicillium lecanii. Doctoral thesis. Universidad Autónoma Metropolitana. Ciudad de México, México, January 23rd, 2015.
- Chen, J.K.; Shen, C.R.; Liu, C.L. N-acetylglucosamine: production and applications. Mar. Drugs. 2010, 8, 2493–2516. [Google Scholar] [CrossRef] [PubMed]
- Faraldos, M.; Goberna, C. Técnicas de análisis y caracterización de materiales, 3rd ed.; Consejo Superior de Investigaciones Científicas: Madrid, Spain, 2021; p. 1052. [Google Scholar]
- Pungor, E.; Horvai, G. A practical guide to instrumental analysis, 1st ed.; CRC press: Boca Raton, London, N.Y., USA, 2020; p. 288. [Google Scholar]
- Dahmane, E.M.; Taourirte, M.; Eladlani, N.; Rhazi, M. Extraction and characterization of chitin and chitosan from Parapenaeus longirostris from Moroccan local sources. Int. J. Polym. Anal. Charact. 2014, 19, 342–351. [Google Scholar] [CrossRef]
- Mohan, K.; Ganesan, A.R.; Muralisankar, T.; Jayakumar, R.; Sathishkumar, P.; Uthayakumar, V.; Revathi, N. Recent insights into the extraction, characterization, and bioactivities of chitin and chitosan from insects. Trends Food Sci. Technol. 2020, 105, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Majtán, J.; Bíliková, K.; Markovič, O.; Gróf, J.; Kogan, G.; Šimúth, J. Isolation and characterization of chitin from bumblebee (Bombus terrestris). Int. J. Biol. Macromol. 2007, 40, 237–241. [Google Scholar] [CrossRef]
- Sáenz-Mendoza, A.; Zamudio-Flores, P.B.; Palomino-Artalejo, G.A.; Tirado-Gallegos, J.M.; García-Cano, V.G.; Ornelas-Paz, J.J.; Aparicio-Saguilán, A. Physicochemical, morphological and structural characterization of the chitin and chitosan of Tenebrio molitor and Galleria mellonella insects. Rev. Mex. Ing. Quim. 2019, 18, 39–56. [Google Scholar] [CrossRef]
- Martínez, C.M.P. Los fundamentos de la cristalografía: una reseña histórica. An. Quím. 2014, 4, 294–302. [Google Scholar]
- Ameh, E.S. A review of basic crystallography and x-ray diffraction applications. Int. J. Adv. Manuf. Technol. 2019, 105, 3289–3302. [Google Scholar] [CrossRef]
- Mohan, K.; Muralisankar, T.; Jayakumar, R.; Rajeevgandhi, C. A study on structural comparisons of α-chitin extracted from marine crustacean shell waste. Carbohydr. Polym. Technol. Appl. 2021, 2, 100037. [Google Scholar] [CrossRef]
- Kaya, M.; Lelešius, E.; Nagrockaitė, R.; Sargin, I.; Arslan, G.; Mol, A.; Bitim, B. Differentiations of chitin content and surface morphologies of chitins extracted from male and female grasshopper species. PloS One. 2015, 10, e0115531. [Google Scholar] [CrossRef] [PubMed]
- Penagos, J.I.C. Caracterización de materiales a través de medidas de microscopía electrónica de barrido (SEM). Elementos 2013, 3, 133–146. [Google Scholar]
- Rojas, J.; Ciro, Y.; Salamanca, C. Efecto del grado de acetilación en las propiedades farmacocinéticas de quitina extraída de exoesqueletos de camarones. Vitae 2018, 25, 87. [Google Scholar]
- Guinesi, L.S.; Cavalheiro, É.T. G. The use of DSC curves to determine the acetylation degree of chitin/chitosan samples. Thermochim. Acta 2006, 444, 128–133. [Google Scholar] [CrossRef]
Figure 1.
Bleaching process showing chitin pre-bleaching treatment (A), chitin after first bleaching treatment (B), and chitin after second bleaching treatment (C).
Figure 1.
Bleaching process showing chitin pre-bleaching treatment (A), chitin after first bleaching treatment (B), and chitin after second bleaching treatment (C).

Figure 5.
SEM images of chitin extracted from Acheta domesticus cricket flour (upper row) with a 970x (a), 5000x (b) and 10000x (c) close-up and of chitosan extracted from Acheta domesticus cricket flour (lower row) with a 1000x (a; b) and 3000x (c) close-up.
Figure 5.
SEM images of chitin extracted from Acheta domesticus cricket flour (upper row) with a 970x (a), 5000x (b) and 10000x (c) close-up and of chitosan extracted from Acheta domesticus cricket flour (lower row) with a 1000x (a; b) and 3000x (c) close-up.

Table 1.
Proximal analysis for Acheta domesticus cricket flour.
| Analysis | Wet base (%) | Dry base (%) | Udomsil et al. [29] (%) |
|---|---|---|---|
| Humidity | 2.55 ± 0.11 | - | 6.3 ± 0.04 |
| Fat | 13.01 ± 0.65 | 13.35 ± 0.65 | 10.4 ± 0.1 |
| Ashes | 4.19 ± 0.04 | 4.30 ± 0.04 | 5.4 ± 0.3 |
| Protein | 54.85 ± 0.69 | 56.29 ± 0.69 | 71.7 ± 0.5 |
| Crude fiber | 15.31 ± 1.01 | 15.71 ± 1.01 | 4.6 ± 0.2 |
| Carbohydrates | 25.40 | 26.06 | 1.6 |
Table 3.
Analysis of variance (ANOVA) for the demineralization process.
| Factor | Levels | Values | P-Value (α = 0.05) |
|---|---|---|---|
| Time (h) | 1 | 3 | 0.230 |
| 2 | 6 | ||
| Temperature (ºC) | 1 | 30 | 0.005 |
| 2 | 60 | ||
| Concentration (M) | 1 | 1 | 0.001 |
| 2 | 2 |
Table 5.
Analysis of variance (ANOVA) for the deproteinization (%DP) process.
| Factor | P-Value (α = 0.05) |
|---|---|
| Temperature (ºC) | 0.119 |
| Time (hrs) | 0.009 |
| Concentration OH (M) | 0.000 |
| Concentration OH * Concentration OH | 0.019 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.


Optimizing Chitin Extraction and Chitosan Production From Domestic Cricket Flour
Andrea Espinosa-Solís
et al.
,
2024
An Uplifting Avenue upon Mealworm Chitosan for Hemodialysis Application
Maria Martingo
et al.
,
2024
Characterization of Farfantepenaeus californiensis Derived Chitosan for Smart-Active Food Packing Applications
Ivone Michel Wong-Miramontes
et al.
,
2024



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated