
Preprint
Review
gRNA Design: How its Evolution Impacted on CRISPR/Cas9 Systems Refinement
Altmetrics
Downloads
274
Views
202
Comments
0
A peer-reviewed article of this preprint also exists.



This version is not peer-reviewed
Submitted:
15 April 2023
Posted:
17 April 2023
You are already at the latest version
Alerts
Abstract
In the last decade, the genetic engineering world has been shaken up by a relatively new genetic editing tool based on RNA-guided Nucleases (RGNs): the CRISPR/Cas9 system. Since the first report in 1987 and its characterization in 2007 as a bacterial defense mechanism, the interest and research on this system have grown exponentially. CRISPR systems provide immunity to bacteria against invading genetic material; however, with specific modifications in sequence and structure, it becomes a precise editing system that makes it possible to genetically modify almost any organism. There are diverse approaches regarding the refinement of these modifications, such as constructing more accurate nucleases, understanding the cellular context and facing the epigenetic conditions, or re-designing guide RNAs (gRNAs). Considering the critical importance for the correct CRISPR/Cas9 systems performance, our scope will emphasize in the latter approach. Hence, we present an overview of the past and the most recent guide RNA web-based design tools, highlighting their computational architecture and gRNA characteristics evolution through the years. Our study concisely explains the computational approaches that use machine learning techniques, deep neural networks, and large datasets of gRNA/target interactions to make possible both predictions and classifications directed to design, optimize, and create promising gRNAs suitable for future gene therapies.
Keywords:
Subject: Biology and Life Sciences - Biology and Biotechnology
1. Introduction
Historically, biotechnology has constantly been improving its techniques and protocols to achieve more straightforward procedures execution and better results [1]. These approaches flowed in small breakthroughs, such as the obtaining of new procedures for purification, amplification, or cleavage; on the other hand, we can mention huge breakthroughs, such as the discovery of bacteria and the first insecticide developed, or even greater, the human genome project [1,2,3,4]. In the case of genome editing or gene insertion methods, a huge variety of machinery has been developed over the years: Regarding plant biotechnology, Ti plasmid-based transformation, founded on Agrobacterium tumefaciens plant infection, has been widely described [5,6], characterized, and compared against different methods with the same objective [7,8]. In a similar way, two methods to perform precisely double-strand DNA breaks (DSBs) were designed; we are explicitly referring to Zinc-Finger Nucleases (ZFN), and Transcription Activator-like Effector Nucleases (TALENs) [9]. Concisely, ZFNs use a dimeric DNA recognition domain fused to fokI restriction enzyme [10,11]. TALENs use the same restriction enzyme, but the dimeric DNA recognition domain from ZFNs is replaced by distinct DNA recognition domains derived from pathogens [12,13]. In addition, both seem to be functional for gene therapy [9,14,15]. Despite the apparent simplicity of these methods, they suffer from strong engineering complexities and limitations [16] that have empowered the pursuit of novel methods to perform even more precise DNA target recognition and cleavage. Thus, genome editing improvement continued in the latter introduced methods. A relatively new programmable tool based on RNA-guided Nucleases (RGNs) has been developed; we are particularly alluding to the type II CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas9 (CRISPR-associated protein 9) genome editing system [16].
1.1. CRISPR Defense System Physiology
The first contact with this system occurred when Ishino et al. [17] described the nucleotide sequence of the iap gene in Escherichia coli in 1987. Until those days, scientists had purely basic and trivial knowledge about this system, and they related it as a cluster of repeated sequences solely spaced by different sequences. In 2007, it was probed that CRISPR/Cas is a defensive bacterial, immunity-providing system against aggressive foreign genetic material such as invading plasmids or bacteriophage DNA [18]. Afterward, the CRISPR/Cas defense system was widely studied and classified into different groups depending on the number of required proteins to protect the respective bacterium. We shall focus on the type II CRISPR/Cas9 system, which involves the obligatory presence of the CRISPR cluster in the bacterial genome, which is based on two principal components (see Figure 1): the CRISPR-associated (Cas) cluster and the CRISPR array. The CRISPR array is formed by an AT-rich leader sequence containing promoter sequences; integrated foreign sequences known as spacers; and palindromic repeats serving as spacer separators [19,20,21,22].
To date, this defense system must complete three stages to provide immunity in the host cell: adaptation, expression, and interference stages [19,23]. Formerly in 2010, these stages were known as the spacer acquisition, processing, and effector stage [24]. The adaptation stage starts as a response of the bacterium to a bacteriophage attack when the insertion of viral genetic material occurs. A host Cas1-Cas2 multimeric protein complex, in association with the Cas9 protein, recognizes the foreign DNA, cuts a specific sequence known as a protospacer, and integrates it into the CRISPR array [20,22]. The expression stage commences during the reinfection by the bacteriophage; the transcript generated from the CRISPR array, pre-CRISPR RNA (pre-crRNA), suffers selective degradation by RNase III to obtain the mature crRNA. The latter forms a double-RNA complex called the crRNA:tracrRNA complex, together with a trans-activating CRISPR RNA (tracrRNA). Next, this complex is bounded to the Cas9 protein in the interference stage, assembling the Cas9:RNA structure [25,26], which performs the target recognition and target degradation activity [23]. Consequently, the Cas9:RNA structure incapacitates the phage from damaging the host (see Figure 2). The target recognition is strongly commanded by a protospacer adjacent motif (PAM) located in the non-target DNA strand, adjacent to the target sequence [25,27,28]. Specific Cas9 protein domains recognize it under the action of essential amino acids [25,28]; however, PAMs do not apport any specificity for Cas9 nuclease domain cleavage [29]. The CRISPR-associated (Cas) proteins are translated from the Cas cluster that is commonly surrounding the neighborhood of the CRISPR array [19]. In 2012, the type II CRISPR/Cas9 system was reprogrammed by its pioneers so it could be used as a genome editing machinery [30,31]. This reprogramming involved the substitution of the crRNA:tracrRNA complex by a synthetic single guide RNA (sgRNA) [31] simplifying the whole system (Figure 3). The term sgRNA (single guide RNA) is completely interchangeable with gRNA (guide RNA). For agility and practical concerns, gRNA will be used in this review.
1.2. gRNAs and CRISPR On-and-Off Targets
There are some important concerns about the gRNA that must be highlighted. First, the common total number of nucleotides that constitute the gRNA recognition site is approximately 20 nucleotides [31,32,33,34], mostly sufficient to have precise target recognition. The seed sequence at the 3’ side of the recognition site plays a major role in the target recognition specificity of the Cas9:gRNA. If there are two or more mismatched nucleotides between the gRNA’s seed sequence and the target sequence, the specificity is notably reduced [28,32]. Mismatches in the PAM-distal positions also reduce the specificity of the Cas9:gRNA, but they are much more tolerated than PAM-surrounding mismatches [35,36].
Even with the apparent simplicity of the gRNA and its efficiency when complexed with the Cas9 protein, there are two fundamental consequences derivated from these circumstances: off-and-on-target bindings. In this context, on-target refers to the ideal hybridization between the gRNA and the target. Off-target bindings are understood as the hybridization between undesired DNA sequences and the gRNA. A high similarity between the gRNA sequence and non-targeting sequences leads to an elevated percentage of these off-target bindings. These off-target effects can be classified depending on the distinct occurrences: Manghwar, Zhang, and Niu [37,38,39] presents three types of off-target effects, regarding "bulges" and simple mismatches; on the other hand, Borrelli et al. [40] present two types, which are much more general and simpler than those from [37,38,39]. Any CRISPR experiment can have off-target bindings and all their adverse and undesired effects. Many strategies have been developed to minimize off-target activity associated with the CRISPR-Cas9 system: from manipulating the proper structure of the Cas9 protein, titration and concentration control of the Cas9 and the gRNA delivered, to altering the gRNA ribonucleotide structure through its in silico designing [34,37,41].
If one highlights the in silico design of gRNAs, a huge variety of patterns and specificities have been described aiming to improve the correct performance of the CRISPR/Cas9 system. For instance, adding extra nucleotides at the 5’ end of the gRNA helps differentiate off-target and on-target sites [32]. Furthermore, the usage of gRNAs whose tracrRNA-fused part is extended provides target cleavage partially improvement [35], or the pinning up of specific nucleotides near the PAM region, in the middle, or at the end of the sequence [33,42,43] gives stability to the gRNA. All of these concerns have been considered when scientists develop algorithms to design optimal gRNAs. Unfortunately, it has been reported that off-target bindings occur even when a considerable amount of nucleotides differ between the gRNA and the off-target site [35,44,45]. Computational areas have been used in the design of these gRNAs with powerful tools such as deep learning [46], which lead to the creation of several models for prediction. The main difference between off-target and on-target CRISPR prediction is the principle: on-target prediction models focus on predicting the effectiveness of the gRNA in cutting a specific target gene. These models identify gRNAs that effectively target a desired gene and are often used in gene editing applications. On the other hand, off-target prediction models, focus on identifying potential unintended effects of a gRNA in cutting other genes in the genome that are not premeditated targets. Both types of prediction models use machine learning techniques, such as neural networks and deep learning, to analyze large datasets of gRNA-target/genome pairs and make predictions about the activity of new gRNAs. From this point, and taking into account that the gRNA is vitally important to perform the recognition and nuclease activity by the Cas9 protein, many efforts were directed to design, optimize, and create better gRNAs. The computational prediction of gRNAs depends on several factors, including the sequence of the CRISPR locus, the genetic context, and the specific algorithm or software being used for the prediction. All these computational approaches shall be explained in detail so the evolution, through time, of the gRNA design is depicted.
2. Machine Learning in gRNA Design
Machine Learning is a branch of artificial intelligence that include algorithms and mathematical models that allow computers to learn from data without being explicitly programmed for each task. The machine learning algorithms follow some steps, starting with data processing, feature extraction, training, and classification or prediction [47].
The input data are DNA sequences that require processing to transform the categorical input into a numerical sequence. The two main algorithms to convert the data to a numeric representation are the "One Hot-Encoding" and "k-mer word embedding" algorithms. One hot encoding is a technique where each value in the vector corresponds to a unique category. Therefore, each base in the gRNA and target DNA can be encoded as one of the four one-hot vectors [1, 0, 0, 0], [0, 1, 0, 0], [0, 0, 1, 0] and [0, 0, 0, 1] [48]. One hot encoding does not capture any information about the relationship between words or the context in which they appear, what the k-mer word embedding algorithm does. However, k-mer word embeddings do not preserve the original sequence of the words and can be sensitive to rare or unseen words.
After the data is processed, it undergoes feature extraction, which involves selecting and transforming the essential characteristics or patterns of the raw data. This selection identifies and converts the most relevant information into a format the machine learning model can understand. The extracted features depend on whether the model is on or off target. Although the same machine learning models can be applied for on and off target prediction (see Table 1 and Table 2), what changes is the interpretation of the data.
During training, a machine learning model searches for patterns in the data. This process also requires setting the hyperparameters, which are variables that control the algorithm’s behavior during learning [47]. For example, the number of layers, neurons, or the type of optimizer. The manipulation of these parameters is essential for enhancing the evaluation metrics of the model.
Machine learning models can output a sequence for prediction tasks or a categorical label for classification tasks. Classification models can be applied to predict if a specific RNA sequence is a potential CRISPR target. Prediction models of CRISPR can forecast the effectiveness of a particular CRISPR-Cas system on a given target sequence. The main machine learning algorithms for predicting or classifying DNA sequences are linear regression algorithms, logistic regression, Decision Trees, Random Forrest, and Support Vector Machines (SVM) (see Figure 4). Neural Networks are another algorithm of artificial intelligence, but due to their complexity, these will be explained in the following section.
Linear regression (LR) is a supervised learning algorithm used for prediction tasks. Linear regression models fit a linear function between the dependent variable and the independent variables. Studies using this algorithm are CRISPRScan [49], CRISPRater [50]. A decision function can be added to a regression model to obtain a logistic regression (LG). In Logistic Regression, a linear function is transformed through a sigmoid function to produce a probability value between 0 and 1, which can then be classified into one of the two categories based on a threshold value. For example, models using this algorithm are Broad GPP [42] and SCC [51].
Decision Trees (DT) is a supervised learning method used for classification and prediction tasks. The algorithm builds a tree-like model of decisions and their possible consequences, where each node represents a feature of the RNA sequence, and each branch represents a possible outcome based on that feature. The algorithm recursively splits the data into subsets based on the most informative features until a stopping criterion is met. An algorithm derived from decision trees is the Random Forest (RF), an ensemble learning algorithm that uses multiple decision trees for classification and prediction tasks. Random forest builds multiple decision trees using randomly selected subsets of the data and features and combines the results of these trees to improve the accuracy of the prediction. Examples of these algorithms are CRISTA [52], Elevation [53] and CHANGE-seq [54]
Support Vector Machines (SVM) are supervised learning algorithms for classification and prediction tasks. SVMs use a similar concept of finding the optimal hyperplane that separates the data points, just like Linear Regression finds the best line that fits the data. However, this algorithm requires a feature extraction and training of the data. The evaluation metrics of this model rely on the significance of the extracted features and the inherent characteristics of the sequence, such as its length or the presence of specific motifs or secondary structures. Examples of studies using SVM are WU-CRISPR [55], SgRNAScorer [56], Azimuth [57], ge-CRISPR [58], and many others.
3. Neural Networks in gRNA Design
Neural Networks (NN) are a branch of machine learning and artificial intelligence that teaches computers to process data in a way inspired by the human brain’s structure and function. It consists of layered networks of neurons that process information and make predictions. Each neuron passes information to its linked neurons by multiplying the input with the link weights and transforming the data with an activation function [47], allowing the network to process complex information. The final layer produces the network’s output, which could be a prediction or a classification of the input data.
NN are trained using a dataset of input-output pairs, where the network learns to map inputs to corresponding outputs by adjusting the weights and biases of its neurons. This process, called backpropagation, involves repeatedly feeding inputs through the network and comparing its output to the desired output, then updating the network’s parameters to minimize the difference between them. This process is repeated for many iterations until the network converges to weights that minimize the loss function. In this way, the NN "learns" to make predictions or classify inputs based on the patterns in the training data.
NN can be designed in various architectures to suit data types and learning tasks. The architecture of a NN is determined by the number and types of layers, the activation functions used, and the connections between the neurons. Different architectures may be more suitable for certain types of data or learning tasks and require different "hyperparameters" to be set to optimize their performance. The most used architectures in the area of CRSPR prediction are the Convolutional Neural Network (CNN) and Recurrent Neural Network (RNN) (see Figure 5). CNN are designed to process two-dimensional images and typically includes convolutional and pooling layers. The main advantage of this architecture is that it combines the process of feature extraction and training in only one process. On the other hand, a RNN are designed to process data sequences and includes recurrent layers such as LSTM or GRU. The main advantage of this architecture is that it can remember previous inputs and use that information for their task.
Recent advances in the field of CRISPR prediction using NN include the development of deep learning-based models that can predict on and off-target effects. These models are trained on large datasets of CRISPR-induced genomic changes and use this information to learn patterns in the data that can be used to predict off-target effects. These models have been used to develop pi-CRISPR (Physically Informed CRISPR), a tool that predicts the potential off-target sites of a gRNA based on its sequence. Another example is the use of convolutional Neural Networks (CNN), a type of deep learning model suited for analyzing DNA sequences in the form of two-dimensional images. These models have been shown to achieve high levels of accuracy in predicting off-target effects and have been used to identify potential off-target sites in the genome that would otherwise have been missed. Recent studies have also focused on developing neural network-based models that can predict on-target effects, such as the activity or potency of a gRNA.
Several other machine learning and deep learning models have been developed for CRISPR prediction, including models for both on-target and off-target prediction. The use of machine learning and deep learning in CRISPR prediction is an active area of research whose improvements have been seen strongly over the years. In the next section, we shall see this evolution until the present time, depicting the architectural changes and experimental research made with these tools.

Table 1.
Off-target models
| Name | Model | Year | Parameter | Detail | Reference |
|---|---|---|---|---|---|
| Experiment | gRNA Optimization | 2013 | NA | [35] | |
| CRISPRtool | SVM | 2013 | R: 0.64 | A library of 73,000 gRNAs was used to generate knockout collections for two human cell lines. | [59] |
| CRISPOR | Self assembled algorithm | 2016 | AUC: 0.91 | [60] | |
| CRISTA | RF | 2017 | Spearman: 0.81. AUROC: 0.96. AUPRC: 0.96. R= 0.8 | GUIDE-Seq, HTGTS, BLESS. | [52] |
| Predict CRISPR | SVM | 2018 | AUROC: 0.99. AUPRC: 0.45 | One hot encoding over Haeussler. | [61] |
| Elevation | GBRT | 2018 | Spearman: 0.98 | One hot encoding over GUIDE-seq. Boench V2 and Haeussler. | [53] |
| DeepCRISPR | DCDNN | 2018 | Spearman: 0.246, AUROC: 0.804, AUPRC: 0.303 | [62] | |
| CNN_std | CNN | 2018 | AUROC: 0.972 | [48] | |
| SynergizingCRISPR | AdaBoost | 2019 | Spearman: 0.938. AUPRC:0.299 | GUIDE-Seq, Haeussler. | [63] |
| sgDesigner | SVM | 2020 | Spearman: 0.750. AUROC: 0.934. Accuracy:0.863 | ||
| CHANGE-seq | GTB | 2020 | AUROC: 0.995,AUPRC: 0.881 | One hot encoding. | [54] |
| CRISPcut | LG, RF, GBT. | 2020 | Accuracy: 0.9149. AUROC: 0.97 | One hot encoding over CIRCLE-seq and CRISPcup. | [64] |
| CRISPR-Net | LRCN | 2020 | AUROC: 0.995. AUPRC: 0.317 | [65] | |
| R-CRISPER | RNN | 2021 | AUROC: 0.991, AUPRC: 0.319 | [39] | |
| piCRISPR | RNN-CNN | 2021 | AUROC: 0.983, AUPRC: 0.978, Spearman: 0.1 | [66] | |
| GCN-CRISPR | 2021 | AUROC: 0.987 | [67] | ||
| CROTON | deep-CNN | 2021 | AUROC: 0.94, AUROC: 0.8112 | [68] | |
| AttCRISPR | Embedding method | 2021 | Spearman: 0.872 | [69] | |
| CRISPR-IP | CNN | 2022 | AUROC: 0.982, Accuracy: 0.990 | [38] |
4. Reaching Efficiency
Since the beginning of the gRNA-design algorithms, scientists have widely used these programs to find the gRNA of interest or gRNAs whose use must be avoided. The concerned efficiency of these programs is of significant importance for research, and it can be measured according to the evaluation metric previously presented. In 2013, Hsu et al. [35] published their web-based off-target sites predictor, CRISPRtool, also known as the MIT CRISPR Design Tool. They designed their experimental data from which they obtained hand-crafted features and implemented a score based on correct matches and mismatches. In 2014, the CRISPRtool was used to design the best gRNAs, targeting two tumor suppressor genes and one oncogene and then mutating them [70] directly for mouse lung cancer; their transient transfection reached a maximum of 44 % of indels. Almost the same results for the work performed by Xue et al. [71] under the same conditions, but for liver cancer in mice. That year, Tsai et al. [72] powered by the use of GUIDE-seq whole-genome sequencing, discovered that the CRISPRtool suffers from the unrecognition of many off-target sites due to very limited parameters implemented in the algorithm.
Approaching 2014, Doench et al. [42] launched the Broad GPP designer, currently relaunched and updated as CRISPick. Machine learning and logistic regression were first used with this on-target prediction engine, releasing new features and updates. Research about genome editing in the parasite Leishmania donovani was performed using CRISPR/Cas9 [73]. Here, the GPP CRISPR designer compared the gRNAa (designed and named by Wei Zhang et al.) with a set of gRNAs suggested by the tool for the gene of interest, resulting in the gRNAa having a low score according to the web-based tool. Additionally, the engine helped to create a robust, high-efficiency protocol to mediate genome editing in Caenorhabditis elegans regardless of possible low-efficiency gRNAs, permitting the use of a wider variety of gRNAs [74].
A gRNA linear regression-based designer model was introduced by Moreno-Mateos et al. [49] in 2015 with CRISPRscan. Thyme et al. [75] found that hairpin formation can reduce gRNA efficiency, and many web-based tools for this purpose before 2016 ignored this critical factor. CRISPRscan was not the exception, but it presented a lower hairpin formation fraction compared with their contenders. In 2016, research about genome modification in hematopoietic stem/progenitor cells (HSPCs) was significantly improved by Gunry et al. [76]. They targeted the CD45 gene in human HL-60 cells with three distinct gRNAs designed with CRISPRscan. Here, high mutagenesis percentages were obtained, touching almost 75 % of indels, which classifies CRISPRscan as a high-fidelity gRNA design tool.
Based on the lack of a model that in different genome contexts widely agglutinates and demonstrates the efficiency provided by distinct sequence features, in 2015, Xu et al. [51] launched the linear regression-based Spacer Scoring for CRISPR (SSC) tool. They aimed to develop an affordable model to design gRNAs for genome-wide functional screens, training it with as many gRNAs datasets as possible for that time. Despite the relatively low ROC-AUC related to its prediction power, Radzisheuskaya et al. [77] utilized this tool to confirm that employing the correct gRNAs, explicitly designed for functional genome screens will highly improve the efficiency, although other factors impact the efficiency strongly. In other words, for CRISPRi (CRISPR gene inhibition), if the gene transcription start site (TTS) is targeted and the highest-scored gRNA for that gene is used, the efficiency will increase, showing better phenotype-based screens.
Plant gRNA prospects and their characteristics partially differ from gRNAs designed for mammals or bacterial cells. For instance, Liang et al. [78] explain that nucleotide preferences in the recognition sequence are not seen for plants, unlike for mammals. Together with the introduction of linear regression-based methods for machine learning training in the last two web-based tools, in 2015, WU-CRISPR and SgRNAScorer [55,56] used the support vector machine (SVM) framework for gRNA design. In contrast to WU-CRISPR, the SgRNAScorer algorithm does not consider the presence of contiguous repetitive sequences, or the impact of RNA’s secondary structures formed in the guide sequence occasioned by self-folding free energy, thus reducing SgRNAScorer efficiency. Even more, Wong et al. compared their WU-CRISPR tool against the SSC, SgRNAScorer, and GPP CRISPR designer tool, demonstrating, using precision-recall curves, a better design of functional gRNAs by WU-CRISPR. Mutagenesis experiments in rice and cotton employed the SgRNAScorer to target genes of interest. In cotton experiments [79], the SgRNAScorer designed 82 distinct gRNAs to target a GFP gene in a transgenic cotton genome, selecting only three significantly different gRNAs in the scoring value. They found that the mutagenesis efficiency varied inconsistently, suggesting that SgRNAScorer gRNA prospects lack robust biological and computational basis. Interestingly, obtaining these results, they decided to use the WU-CRISPR tool, getting only 13 gRNAs for their gene. Analogously, in rice experiments, Baysal et al. [80] selected two gRNAs for a gene of interest. Unfortunately and inconsistently, the high-scored gRNA showed no mutagenesis activity, whereas the lowest one positively did.
Recalling the Broad GPP designer by Doench et al. [42], whose architecture was based on the support vector machine (SVM) with logistic regression, in 2016, it was improved by the launch of Azimuth [57]. This tool seeks the integration of biochemical and thermodynamic sequence features regarding the secondary structure formation, a characteristic missing in the first version of this tool. In addition, they found a better performance and incorporated linear regression models, specifically gradient-boosted regression trees, which proved to be much more efficient than the first version. Finally, they provided two score-based parameters for accurately discriminating potential on-target and off-target sites: Rule Set 2, and the CFD score, respectively, incorporated in their Azimuth web page. Two years later, Listgarten and colleagues developed the Elevation tool [53], an off-target-prediction-focused algorithm that aims to complement the Azimuth model, changing the architecture for a two-layer stacked regression model, where the first layer is intended to learn to predict unique mismatches in the gRNA-target duplexes. The second layer learns to predict various mismatches, yielding a score for potential off-target sites.
As explained throughout this section, the off-target predictor, CRISPRtool, by Hsu et al. [35] suffered from many weaknesses, invoking the necessity of a potent tool to predict off-target sites. In 2016, Haeussler et al. [60] launched the off-target predictor CRISPOR, powered by the BWA sequence search algorithm [81] to perform the corresponding alignments to locate possible off-target sites. CRISPOR’s predicted gRNAs avoid using extremely GC-rich sequences, and the tool treats >4 mismatches much better than the MIT CRISPRtool. These patterns found by analyzing eight large datasets of off-target sites deliver an improved fidelity on CRISPOR prediction. Mutagenesis and gene knock-out research in the hexaploid Camelina sativa [82] employed the CRISPOR tool to design desired and exclude undesired gRNAs for targeting the microsomal oleate desaturase (FAD2) gene, whose knock-out leads to an accumulation of oleic acid in this plant. They selected two gRNAs, from which the second one harbors sequence features described by CRISPOR to improve the mutagenesis efficiency. Looking back on the sgRNA Scorer, Chari et al. [56] structured this tool to analyze gRNA sets of high and low activity for two orthologs of the Cas9 protein. For each ortholog, a separate SVM model was created. In 2017, the same team founded the sgRNA Scorer 2.0 [84], which inversely creates just one SVM model for both Cas9 orthologs by merging all gRNAs in high and low activity sets. With this, they aimed to design a model that predicts efficient gRNAs for distinct CRISPR systems, knowing that many orthologs exist for different CRISPR systems. Even though this tool was trained with a dataset of gRNAs targeting eukaryotic cell genes, Shen et al. [98] used this tool to design 81 gRNAs targeting virulent Klebsiella phage genes. As expected, due to the cellular context, sgRNAScorer did not discriminate correctly between high-and-low-activity gRNAs.
In 2016, the research done for CRISPOR’s feature incorporation shall cause inconsistencies with the research by Abadi et al. [52]. The latter team launched in 2017 a new predictor known as CRISTA (CRISPR Target Assessment), based on a regression model using the Random Forest algorithm. Their primary purpose was not to design a model for exclusively predicting gRNA on-target efficiency or potential off-target sites but to assess the cleavage efficacy of a particular genomic target by a specific gRNA. CRISTA included a treatment for DNA/RNA “bulges” in their algorithm, which can be understood as gaps in the gRNA/target hybridization. CRISPOR noticed these bulges, but their database analysis suggested no need for treating these gaps, disfavoring this tool for missing this important feature. CRISTA finally considered the necessity to deal with the formation of secondary structures inherent to RNA sequences by their learning model. Furthermore, the DNA enthalpy, geometry, and the target location (chromosome number and distance from telomere and centromere) are some additional features inserted in the algorithm. In contrast to many other predicting tools, the CRISTA training dataset does not discard uncleaved sites (i.e., targeted sequences with no gRNA activity), helping to avoid the design of identical zero-activity gRNAs.
Table 2.
On-target models
| Name | Model | Year | Parameter | Detail | Reference |
|---|---|---|---|---|---|
| Broad GPP | LG | 2014 | Spearman: 0.87 | 1,831gRNAs targeting three human genes and six mouse genes were used to generate screening data using one-hot encoding | [42] |
| WU-CRISPR | SVM | 2015 | AUROC 0.91, Spearman 0.70 | [55] | |
| SSC | LG | 2015 | AUROC : 0.711 | Datasets Wang, Koik Yusa, Shalcm, Zhou, Gilbert, Konermann. | [51] |
| Multiple CRISPR models | SVM, LR, GBT, LG, RF | 2015 | Spearman : 0.51. AUROC : 0.75 | One hot encoding over the datasets Wang ribosomal, Wang non-ribosomal, Koike-Yusa, Doench Vl. | [83] |
| CRISPRScan | LR | 2015 | R: 0.45, SD: 0.071 | Includes data from new cell lines. | [49] |
| SgRNAScorer | SVM | 2015 | Spearman 0.75 | [56] | |
| Azimuth | SVM, LG | 2016 | 0.462 | One hot encoding. | [57] |
| ge-CRISPR | SVM | 2016 | Accuracy: 0.888. MCC: 0.78 | Includes data from new cell lines. | [58] |
| CRISPRater | LR | 2017 | Spearman 0.67 | Includes data from new cell lines. | [50] |
| SgRNAScorer 2.0 | SVM | 2017 | Accuracy: 0.737, Precision: 0.728, Recall of 0.758 | [84] | |
| CRISPRpred | SVM | 2017 | AUROC: 0.85. AUPRC: 0.56. MCC: 0.4 | K-mer encoding over Broad GPP. | [85] |
| DeepCRISPR | CNN | 2018 | Spearman 0.406 | [62] | |
| DeepCpf1 | CNN | 2018 | Spearman:0.873 | [86] | |
| DeepCas9 | CNN | 2018 | Spearman 0.351 | [87] | |
| TUSCAN | RF | 2018 | Spearman: 0.55 | [88] | |
| DeepHF | RNN | 2019 | Spearman: 0.867 | Cell lines HCT116, HEK293T, HELA, HL60. | [89] |
| DeepSpCas9 | 1DCNN | 2019 | Spearman: 0.91 | [90] | |
| CRISPRpred(SEQ) | SVM | 2020 | Spearman: 0.829. AUROC: 0.893 | Haeussler and DeepHF datasets. | [91] |
| GNL-Scorer | AdaBoost | 2020 | Spearman: 0.502 | One hot encoding over 10 public datasets. | [92] |
| C-RNN CRISPR | RNN | 2020 | Spearman: 0.877. AUROC: 0.976 | Includes data from new cell lines. | [93] |
| CNN-SVR CRISPR | CNN-SVR | 2020 | Spearman: 0.807. AUROC: 0.983 | Includes data from new cell lines. | [94] |
| On-target CRISPRon | CNN | 2021 | Spearman 0.91 | [95] | |
| BoostMEC | GBM | 2022 | 0.704 | Includes data from new cell lines. | [96] |
| CNN-XG | CNN-Tree | 2022 | Spearman 0.7352 AUROC: 0.992 | [97] |
The CRISPR/Cas9 genome editing system left the scientific community with gigantic expectations. The promise of flawless gene knock-out, knock-in, or functional screens must be accomplished. In 2018, Chuai et al. [62] finally included the use of deep neural network approaches for predicting and designing gRNAs into their novel tool, DeepCRISPR. Parallelly to CRISTA, DeepCRISPR seeks to predict both functional on-target gRNAs and avoid those with a propensity to rise off-target cuts.
Figure 7.
Timeline from 2018 to 2022. With the launching of DeepCRISPR, deep neuronal networks initiated its treasure, improving each year with the introduction of RNN, embedding methods, hybrid models or addition of more layers.
Figure 7.
Timeline from 2018 to 2022. With the launching of DeepCRISPR, deep neuronal networks initiated its treasure, improving each year with the introduction of RNN, embedding methods, hybrid models or addition of more layers.

In order to achieve this purpose, Chuai et al. designed an architecture with three fundamental networks: the main one can be understood as the pre-training network (known as “parent network” by the authors) that will recognize various features of gRNAs, using as input ∼ 0.68 billion gRNA sequences targeting coding and non-coding human genes. The following two CNN use the pre-training network corresponding output. These last networks are trained using well-known, experimentally validated gRNAs with on-target or off-target activity, extracting all the distinctive features characterizing these sequences for further integration in the predictive capacity of the tool. In 2020, accordingly to the pre-training DeepCRISPR dataset based on human exons and intron genes, the tool helped to predict the off-target activity of gRNAs designed by Mintz et al. [99] that initially targets the PARP1 gene, for its inhibition, in triple-negative breast cancer (TNBC) cells, highlighting the importance of using CRISPR/Cas9 systems in preclinical studies.
In the same year of the DeepCRISPR launch, Lin et al. [48] focused on developing a tool that exclusively predicts off-target sites with a deep neural network framework. They named their tool as CNN_std, in which they adapted the biological ribonucleotide sequence of the gRNA for the computational environment in a matrix with 4 x 23 size, representing the four nucleotides and the 20-nt recognition sequence plus the 3-nt PAM sequence. This matrix has the correct format for input in the convolutional neural network. Also, Lin et al. utilized the CRISPOR dataset for training, validating, testing, and comparing CNN_std against previous off-target prediction tools such as the CFD score or the MIT CRISPR design tool, overperforming all these and other machine learning-based tools getting a ROC-AUC of 0.972.
Undoubtedly, the CRISPR/Cas9 systems had an enormous refinement with the introduction of deep neural networks, specifically CNN. Unluckily, DeepCRISPR and CNN_std implemented algorithms and architectures that neglected the biological features underlying the gRNA feasible design, thus missing characteristics probed to be crucial for this objective. Also, the Chari et al. sgRNA Scorer, the first and second version [56,84], used an algorithm trained with datasets obtained from diverse Cas9 orthologs, then being capable of predicting under a more comprehensive array of RGNs (RNA-guided nucleases). In 2019, Wang et al. [89] compared the predicting performance of an RNN and a conventional CNN. They found that RNN beats CNN and other machine learning algorithms. The dataset used for training, validation, and testing is based on their own experiments in human cells, emphasizing the use of three Cas9 orthologs: WT-SpCas9 (wild-type Streptococcus pyogenes Cas9), eSpCas9 (enhanced), and SpCas9-HF1 (High Fidelity). Furthermore, to remedy the inexistence of biological features treatment in deep neural network models, this RNN was trained with features such as sequence secondary structure formation and their stem-loops, GC content, or the contiguous repetitive sequences first described by Wong et al. [55], and implemented in the WU-CRISPR tool in 2015. Lastly, Wang and his team launched DeepHF, a tool comprising all the concerns mentioned earlier. DeepHF was used in experiments premeditated to knock out an apoptosis-inducing gene in mice, Htra2, whose translated protein is found in high concentrations in neomycin-treated cochleae, one of the causes to develop deafness. The team designed three gRNAs targeting the Htra2 gene, obtaining 87.27 % of indels in the Htra2 gene for the highest-scored gRNA [100].
Notwithstanding the boom of deep learning-based pipelines in gRNA design tools, Muhammad et al. [91] were uncomfortable using deep neural networks for gRNA design. Despite the visible characteristics and performance obtained with these models (CNN or RNN), it is tough to interpret their results. Even more, it has been proven that conventional, simpler algorithms can perform the same work done by deep neural networks [101]. Regarding the latter point, Muhammad et al. launched the on-target CRISPRpred(SEQ) predictor tool, whose SVM-based architecture was trained with the same training dataset for DeepCRISPR while mixing biological gRNA sequence features. In most of the benchmarks, CRISPRpred(SEQ) outperformed DeepCRISPR. On the other hand, CRISPRpred(SEQ) challenged DeepHF using the dataset generated by the latter; unluckily, the machine learning-based tool did not surpass DeepHF due to needing more specific tuning against DeepHF.
Another scope to achieve the desired interpretability in deep neural networks is presented by Xiao et al. [69]. Firstly, they provide a categorization of the existing deep neuronal networks, founded on the treatment of the model’s input: methods in the spatial domain, whose input is transformed in a two-dimensional image, which is ready to work with convolutional neural networks for sequence feature extraction [48,62]; methods in the temporal domain, for which the input is treated as a word, and works perfectly with recurrent convolutional networks [89]. Xiao et al. then proposed an ensemble learning model that uses both the spatial and temporal domains to extract the necessary sequence features, in addition to an “attention mechanism” to give interpretability. The on-target model, which is named AttCRISPR, was further enhanced with hand-crafted biological features, finally overperforming even the DeepHF tool with its training dataset.
In recent years, almost all gRNA design tools have turned their vision to implement only deep neural networks, or hybrid models. These models are increasingly perfecting the predictive activity, getting more and more computationally flawless. In 2022, Zhang et al. [38] launched the off-target CRISPR-IP predictor tool, which includes four layers, each of which performs distinct procedures focused on characterizing novel sequence features; these are CNN, Bi-directional Long-Short Term Memory (BiLSTM, an RNN derivative), attention layer, and finally a dense layer. The model uses as training dataset experimental information based on sequencing (SITE-seq and CIRCLE-seq). Finally, epigenetic information and bulge treatment were adapted to the model, resulting in high predicting performance. Regarding the most recent on-target prediction tool, Li et al. [97] proposed a machine-and-deep learning hybrid model. They got inspiration from a fully-computational approach published by Ren et al. [102] that seeks to provide an accurate and high-performance image classification based on XGBoost (extreme gradient-boosted tree, being the machine learning part) and CNN (the deep learning and feature extraction part). The computational approach thus was fused with the biological vision in the hybrid model named CNN-XG, using as input a gRNA sequence, treating it with the CNN layer for feature extraction, and finally sending the latter as an input for the XGBoost classification structure.
5. Conclusions and Future Directions
Over the years, computational approaches have been implemented to design highly accurate single-guide RNAs. The increasing implementation of CRISPR/Cas9 systems for gene editing motivated the improvement of new tools to reduce off-target effects. From the first non-learning algorithm to the use of complex multiple-layer or hybrid machine-and-deep learning architectures, vast computational and biological features are underlying and supporting the advances of CRISPR/Cas9 in gene therapy, or in vivo genome editing. Despite the high scores obtained by deep neural networks, they suffer from low interpretability, making computational and biological scientists confide again in machine learning models, as occurred in 2020 with CRISPRpred(SEQ) [91]. Thus, deep neural network models fitted machine learning structures as part of their architecture to provide users with a powerful and minimalist tool. As [97] presented in their CNN-XG tool, hybrid frameworks for the gRNA design seem to be really feasible to surpass all past architectures, while providing the best features of learning-based algorithms. Consequently, the assembly of hybrid models including approaches from deep neural networks and machine learning must be investigated in depth.
The challenges for future research in sgRNA design are enormous. Among others, computational models should focus on tuning the hyperparameters that appear in the architecture design to increase the model’s user interpretability. It is of major relevance to include in the model biological features found in the laboratory and in silico. Thus, for the next steps in gRNA design, many more CRISPR/Cas9 activity datasets are required to address biological and epigenetic concerns: the more data from different human and plant cell lines, and unicellular organisms, the more biological functional features found.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Niazi, S.K. Handbook of Biogeneric Therapeutic Proteins; Taylor & Francis Group: New York, 2006; p. 585. [Google Scholar] [CrossRef]
- Cantor, C.R.; Smith, C. Genomics - The Science and Technology Behind the Human Genome Project; Vol. 2, John Wiley & Sons, Inc, 1999; pp. 206–208. [CrossRef]
- Caplan, A and Claes, B and Dekeyser, R and Van Montagu, M. Current Plant Science and Biotechnology in Agriculture, 1 ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1990; pp. 90–247. [Google Scholar] [CrossRef]
- Richards, J.E. The Human Genome - A User’s Guide, 2 ed.; Elsevier Academic Press: Burlington, USA, 2005; p. 478. [Google Scholar]
- Zhu, J.; Oger, P.M.; Schrammeijer, B.; Hooykaas, P.J.; Farrand, S.K.; Winans, S.C. The bases of crown gall tumorigenesis. Journal of Bacteriology 2000, 182, 3885–3895. [Google Scholar] [CrossRef] [PubMed]
- Zupan, J.; Muth, T.R.; Draper, O.; Zambryski, P. The transfer of DNA from Agrobacterium tumefaciens into plants: A feast of fundamental insights. Plant Journal 2000, 23, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Weyda, I.; Yang, L.; Vang, J.; Ahring, B.K.; Lübeck, M.; Lübeck, P.S. A comparison of Agrobacterium-mediated transformation and protoplast-mediated transformation with CRISPR-Cas9 and bipartite gene targeting substrates, as effective gene targeting tools for Aspergillus carbonarius. Journal of Microbiological Methods 2017, 135, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Michielse, C.B.; Arentshorst, M.; Ram, A.F.; Van Den Hondel, C.A. Agrobacterium-mediated transformation leads to improved gene replacement efficiency in Aspergillus awamori. Fungal Genetics and Biology 2005, 42, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Klug, W.S.; Cummings, M.R. Concepts of Genetics, 20 ed.; Vol. 48, Pearson Education, Inc.: Hoboken, New Jersey, 2019; p. 867. [Google Scholar]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proceedings of the National Academy of Sciences of the United States of America 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Cathomen, T.; Keith Joung, J. Zinc-finger nucleases: The next generation emerges. Molecular Therapy 2008, 16, 1200–1207. [Google Scholar] [CrossRef]
- Moscou, M.J.; Bogdanove, A.J. Recognition by TAL Effectors. Science (New York, N.Y.) 2009, 326, 1501. [Google Scholar] [CrossRef]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 756–761. [Google Scholar] [CrossRef]
- Ding, Q.; Lee, Y.K.; Schaefer, E.A.; Peters, D.T.; Veres, A.; Kim, K.; Kuperwasser, N.; Motola, D.L.; Meissner, T.B.; Hendriks, W.T.; et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell 2013, 12, 238–251. [Google Scholar] [CrossRef]
- Benjamin, R.; Berges, B.K.; Solis-Leal, A.; Igbinedion, O.; Strong, C.L.; Schiller, M.R. TALEN gene editing takes aim on HIV. Human Genetics 2016, 135, 1059–1070. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakatura, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isoenzyme conversion in Escherichia coli, and identification of the gene product. Journal of Bacteriology 1987, 169, 5429–5433. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Mohamadi, S.; Bostanabad, S.Z.; Mirnejad, R. CRISPR arrays: A review on its mechanism. Journal of Applied Biotechnology Reports 2020, 7, 81–86. [Google Scholar] [CrossRef]
- Wright, A.V.; Liu, J.J.; Knott, G.J.; Doxzen, K.W.; Nogales, E.; Doudna, J.A. Structures of the CRISPR genome integration complex. Science 2017, 357, 1113–1118. [Google Scholar] [CrossRef]
- Nuñez, J.K.; Lee, A.S.; Engelman, A.; Doudna, J.A. Integrase-mediated spacer acquisition during CRISPR-Cas adaptive immunity. Nature 2015, 519, 193–198. [Google Scholar] [CrossRef]
- Xiao, Y.; Ng, S.; Hyun Nam, K.; Ke, A. How type II CRISPR-Cas establish immunity through Cas1-Cas2-mediated spacer integration. Nature 2017, 550, 137–141. [Google Scholar] [CrossRef]
- Rath, D.; Amlinger, L.; Rath, A.; Lundgren, M. The CRISPR-Cas immune system: Biology, mechanisms and applications. Biochimie 2015, 117, 119–128. [Google Scholar] [CrossRef]
- Karginov, F.V.; Hannon, G.J. The CRISPR System: Small RNA-Guided Defense in Bacteria and Archaea. Molecular Cell 2010, 37, 7–19. [Google Scholar] [CrossRef]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014, 343. [Google Scholar] [CrossRef]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhou, K.; Ma, L.; Gressel, S.; Doudna, J.A. A Cas9-guide RNA complex preorganized for target DNA recognition. Science 2015, 348, 1477–1481. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Erdmann, S.; Mojica, F.J.; Garrett, R.A. Protospacer recognition motifs: Mixed identities and functional diversity. RNA Biology 2013, 10, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, H.; Chen, L.L.; Xie, K. Recent advances in genome editing using CRISPR/Cas9. Frontiers in Plant Science 2016, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA – Guided. Science 2012, 337, 816–822. [Google Scholar] [CrossRef]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Research 2013, 24, 132–141. [Google Scholar] [CrossRef]
- Wu, X.; Kriz, A.J.; Sharp, P.A. Target specificity of the CRISPR-Cas9 system. Quantitative Biology 2014, 2, 59–70. [Google Scholar] [CrossRef]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Molecular Therapy - Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology 2013, 31, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Manghwar, H.; Li, B.; Ding, X.; Hussain, A.; Lindsey, K.; Zhang, X.; Jin, S. CRISPR/Cas Systems in Genome Editing: Methodologies and Tools for sgRNA Design, Off-Target Evaluation, and Strategies to Mitigate Off-Target Effects. Advanced Science 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.R.; Jiang, Z.R. Effective use of sequence information to predict CRISPR-Cas9 off-target. Computational and Structural Biotechnology Journal 2022, 20, 650–661. [Google Scholar] [CrossRef]
- Niu, R.; Peng, J.; Zhang, Z.; Shang, X. R-CRISPR: A deep learning network to predict off-target activities with mismatch, insertion and deletion in CRISPR-Cas9 system. Genes 2021, 12. [Google Scholar] [CrossRef]
- Borrelli, V.M.; Brambilla, V.; Rogowsky, P.; Marocco, A.; Lanubile, A. The enhancement of plant disease resistance using crispr/cas9 technology. Frontiers in Plant Science 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nature Biotechnology 2014, 32, 279–284. [Google Scholar] [CrossRef]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology 2014, 32, 1262–1267. [Google Scholar] [CrossRef]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology 2013, 31, 822–826. [Google Scholar] [CrossRef]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nature Biotechnology 2013, 31, 839–843. [Google Scholar] [CrossRef]
- Konstantakos, V.; Nentidis, A.; Krithara, A.; Paliouras, G. CRISPR-Cas9 gRNA efficiency prediction: an overview of predictive tools and the role of deep learning. Nucleic Acids Research 2022, 50, 3616–3637. [Google Scholar] [CrossRef]
- Volk, M.J.; Lourentzou, I.; Mishra, S.; Vo, L.T.; Zhai, C.; Zhao, H. Biosystems Design by Machine Learning. ACS Synthetic Biology 2020, 9. [Google Scholar] [CrossRef]
- Lin, J.; Wong, K.C. Off-target predictions in CRISPR-Cas9 gene editing using deep learning. 2018, Vol. 34. [CrossRef]
- Moreno-Mateos, M.A.; Vejnar, C.E.; Beaudoin, J.D.; Fernandez, J.P.; Mis, E.K.; Khokha, M.K.; Giraldez, A.J. CRISPRscan: Designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nature Methods 2015, 12. [Google Scholar] [CrossRef]
- Labuhn, M.; Adams, F.F.; Ng, M.; Knoess, S.; Schambach, A.; Charpentier, E.M.; Schwarzer, A.; Mateo, J.L.; Klusmann, J.H.; Heckl, D. Refined sgRNA efficacy prediction improves largeand small-scale CRISPR-Cas9 applications. Nucleic Acids Research 2018, 46. [Google Scholar] [CrossRef]
- Xu, H.; Xiao, T.; Chen, C.H.; Li, W.; Meyer, C.A.; Wu, Q.; Wu, D.; Cong, L.; Zhang, F.; Liu, J.S.; et al. Sequence determinants of improved CRISPR sgRNA design. Genome Research 2015, 25, 1147–1157. [Google Scholar] [CrossRef]
- Abadi, S.; Yan, W.X.; Amar, D.; Mayrose, I. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Computational Biology 2017, 13. [Google Scholar] [CrossRef]
- Listgarten, J.; Weinstein, M.; Kleinstiver, B.P.; Sousa, A.A.; Joung, J.K.; Crawford, J.; Gao, K.; Hoang, L.; Elibol, M.; Doench, J.G.; et al. Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nature Biomedical Engineering 2018, 2. [Google Scholar] [CrossRef] [PubMed]
- Lazzarotto, C.R.; Malinin, N.L.; Li, Y.; Zhang, R.; Yang, Y.; Lee, G.H.; Cowley, E.; He, Y.; Lan, X.; Jividen, K.; et al. CHANGE-seq reveals genetic and epigenetic effects on CRISPR–Cas9 genome-wide activity. Nature Biotechnology 2020, 38. [Google Scholar] [CrossRef]
- Wong, N.; Liu, W.; Wang, X. WU-CRISPR: characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome biology 2015, 16, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.; Mali, P.; Moosburner, M.; Church, G.M. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nature Methods 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology 2016, 34, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Gupta, A.K.; Rajput, A.; Kumar, M. Ge-CRISPR - An integrated pipeline for the prediction and analysis of sgRNAs genome editing efficiency for CRISPR/Cas system. Scientific Reports 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2013, 343, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, M.; Schönig, K.; Eckert, H.; Eschstruth, A.; Mianné, J.; Renaud, J.B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology 2016, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Zheng, Y.; Zhao, Z.; Liu, T.; Li, J. Recognition of CRISPR/Cas9 off-target sites through ensemble learning of uneven mismatch distributions. 2018, Vol. 34. [CrossRef]
- Chuai, G.; Ma, H.; Yan, J.; Chen, M.; Hong, N.; Xue, D.; Zhou, C.; Zhu, C.; Chen, K.; Duan, B.; et al. DeepCRISPR: Optimized CRISPR guide RNA design by deep learning. Genome Biology 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, X.; Lin, Q.; Wong, K.C. Synergizing CRISPR/Cas9 off-target predictions for ensemble insights and practical applications. Bioinformatics 2019, 35. [Google Scholar] [CrossRef]
- Dhanjal, J.K.; Dammalapati, S.; Pal, S.; Sundar, D. Evaluation of off-targets predicted by sgRNA design tools. Genomics 2020, 112. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, Z.; Zhang, S.; Chen, J.; Wong, K.C. CRISPR-Net: A Recurrent Convolutional Network Quantifies CRISPR Off-Target Activities with Mismatches and Indels. Advanced Science 2020, 7. [Google Scholar] [CrossRef]
- Störtz, F.; Mak, J.; Minary, P. piCRISPR: Physically Informed Features Improve Deep Learning Models for CRISPR/Cas9 Off-Target Cleavage Prediction. bioRxiv 2021. [Google Scholar] [CrossRef]
- Vinodkumar, P.K.; Ozcinar, C.; Anbarjafari, G. Prediction of sgRNA off-target activity in CRISPR/Cas9 gene editing using graph convolution network. Entropy 2021, 23. [Google Scholar] [CrossRef]
- Li, V.R.; Zhang, Z.; Troyanskaya, O.G. CROTON: An automated and variant-aware deep learning framework for predicting CRISPR/Cas9 editing outcomes. Bioinformatics 2021, 37. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.M.; Wan, Y.Q.; Jiang, Z.R. AttCRISPR: a spacetime interpretable model for prediction of sgRNA on-target activity. BMC Bioinformatics 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Chen, S.; Yin, H.; Tammela, T.; Papagiannakopoulos, T.; Joshi, N.S.; Cai, W.; Yang, G.; Bronson, R.; Crowley, D.G.; et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 2014, 514, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature biotechnology 2015, 33, 187–197. [Google Scholar] [CrossRef]
- Zhang, W.W.; Matlashewski, G. CRISPR-Cas9-mediated genome editing in Leishmania donovani. MBio 2015, 6, e00861–15. [Google Scholar] [CrossRef]
- Paix, A.; Folkmann, A.; Rasoloson, D.; Seydoux, G. High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9ribonucleoprotein complexes. Genetics 2015, 201, 47–54. [Google Scholar] [CrossRef]
- Thyme, S.B.; Akhmetova, L.; Montague, T.G.; Valen, E.; Schier, A.F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nature Communications 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Gundry, M.C.; Brunetti, L.; Lin, A.; Mayle, A.E.; Kitano, A.; Wagner, D.; Hsu, J.I.; Hoegenauer, K.A.; Rooney, C.M.; Goodell, M.A.; et al. Highly Efficient Genome Editing of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. Cell Reports 2016, 17, 1453–1461. [Google Scholar] [CrossRef]
- Radzisheuskaya, A.; Shlyueva, D.; Müller, I.; Helin, K. Optimizing sgRNA position markedly improves the efficiency of CRISPR/dCas9-mediated transcriptional repression. Nucleic acids research 2016, 44, e141–e141. [Google Scholar] [CrossRef]
- Liang, G.; Zhang, H.; Lou, D.; Yu, D. Selection of highly efficient sgRNAs for CRISPR/Cas9-based plant genome editing. Scientific reports 2016, 6, 21451. [Google Scholar] [CrossRef] [PubMed]
- Janga, M.R.; Campbell, L.M.; Rathore, K.S. CRISPR/Cas9-mediated targeted mutagenesis in upland cotton (Gossypium hirsutum L.). Plant Molecular Biology 2017, 94, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Baysal, C.; Bortesi, L.; Zhu, C.; Farré, G.; Schillberg, S.; Christou, P. CRISPR/Cas9 activity in the rice OsBEIIb gene does not induce off-target effects in the closely related paralog OsBEIIa. Molecular Breeding 2016, 36, 1–11. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Morineau, C.; Bellec, Y.; Tellier, F.; Gissot, L.; Kelemen, Z.; Nogué, F.; Faure, J.D. Selective gene dosage by CRISPR-Cas9 genome editing in hexaploid Camelina sativa. Plant biotechnology journal 2017, 15, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Fusi, N.; Smith, I.; Doench, J.; Listgarten, J. In Silico Predictive Modeling of CRISPR/Cas9 guide efficiency. bioRxiv 2015. [Google Scholar] [CrossRef]
- Chari, R.; Yeo, N.C.; Chavez, A.; Church, G.M. SgRNA Scorer 2.0: A Species-Independent Model to Predict CRISPR/Cas9 Activity. ACS Synthetic Biology 2017, 6. [Google Scholar] [CrossRef]
- Rahman, M.K.; Rahman, M.S. CRISPRpred: A flexible and efficient tool for sgRNAs on-target activity prediction in CRISPR/Cas9 systems. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Kim, H.K.; Min, S.; Song, M.; Jung, S.; Choi, J.W.; Kim, Y.; Lee, S.; Yoon, S.; Kim, H. Deep learning improves prediction of CRISPR-Cpf1 guide RNA activity. Nature Biotechnology 2018, 36. [Google Scholar] [CrossRef]
- Xue, L.; Tang, B.; Chen, W.; Luo, J. Prediction of CRISPR sgRNA Activity Using a Deep Convolutional Neural Network. Journal of Chemical Information and Modeling 2019, 59. [Google Scholar] [CrossRef]
- Wilson, L.O.; Reti, D.; O’Brien, A.R.; Dunne, R.A.; Bauer, D.C. High activity target-site identification using phenotypic independent CRISPR-Cas9 core functionality. The CRISPR Journal 2018, 1, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, C.; Wang, B.; Li, B.; Wang, Q.; Liu, D.; Wang, H.; Zhou, Y.; Shi, L.; Lan, F.; et al. Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nature Communications 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Kim, Y.; Lee, S.; Min, S.; Bae, J.Y.; Choi, J.W.; Park, J.; Jung, D.; Yoon, S.; Kim, H.H. SpCas9 activity prediction by DeepSpCas9, a deep learning–based model with high generalization performance. Science advances 2019, 5, eaax9249. [Google Scholar] [CrossRef] [PubMed]
- Rafid, A.H.M.; Toufikuzzaman, M.; Rahman, M.S.; Rahman, M.S. CRISPRpred(SEQ): A sequence-based method for sgRNA on target activity prediction using traditional machine learning. BMC Bioinformatics 2020, 21. [Google Scholar] [CrossRef]
- Wang, J.; Xiang, X.; Bolund, L.; Zhang, X.; Cheng, L.; Luo, Y. GNL-Scorer: A generalized model for predicting CRISPR on-target activity by machine learning and featurization. Journal of Molecular Cell Biology 2020, 12. [Google Scholar] [CrossRef]
- Zhang, G.; Dai, Z.; Dai, X. C-RNNCrispr: Prediction of CRISPR/Cas9 sgRNA activity using convolutional and recurrent neural networks. Computational and Structural Biotechnology Journal 2020, 18. [Google Scholar] [CrossRef]
- Zhang, G.; Dai, Z.; Dai, X. A Novel Hybrid CNN-SVR for CRISPR/Cas9 Guide RNA Activity Prediction. Frontiers in Genetics 2020, 10. [Google Scholar] [CrossRef]
- Xiang, X.; Corsi, G.I.; Anthon, C.; Qu, K.; Pan, X.; Liang, X.; Han, P.; Dong, Z.; Liu, L.; Zhong, J.; et al. Enhancing CRISPR-Cas9 gRNA efficiency prediction by data integration and deep learning. Nature Communications 2021, 12. [Google Scholar] [CrossRef]
- Zarate, O.A.; Yang, Y.; Wang, X.; Wang, J.P. BoostMEC: predicting CRISPR-Cas9 cleavage efficiency through boosting models. BMC Bioinformatics 2022, 23, 446. [Google Scholar] [CrossRef]
- Li, B.; Ai, D.; Liu, X. CNN-XG: A Hybrid Framework for sgRNA On-Target Prediction. Biomolecules 2022, 12. [Google Scholar] [CrossRef]
- Shen, J.; Zhou, J.; Chen, G.Q.; Xiu, Z.L. Efficient genome engineering of a virulent Klebsiella bacteriophage using CRISPR-Cas9. Journal of Virology 2018, 92, e00534–18. [Google Scholar] [CrossRef] [PubMed]
- Mintz, R.L.; Lao, Y.H.; Chi, C.W.; He, S.; Li, M.; Quek, C.H.; Shao, D.; Chen, B.; Han, J.; Wang, S.; et al. CRISPR/Cas9-mediated mutagenesis to validate the synergy between PARP1 inhibition and chemotherapy in BRCA1-mutated breast cancer cells. Bioengineering & Translational Medicine 2020, 5, e10152. [Google Scholar] [CrossRef]
- Gu, X.; Wang, D.; Xu, Z.; Wang, J.; Guo, L.; Chai, R.; Li, G.; Shu, Y.; Li, H. Prevention of acquired sensorineural hearing loss in mice by in vivo Htra2 gene editing. Genome Biology 2021, 22, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Ferrari Dacrema, M.; Cremonesi, P.; Jannach, D. Are we really making much progress? A worrying analysis of recent neural recommendation approaches. In Proceedings of the Proceedings of the 13th ACM conference on recommender systems, 2019, pp.; pp. 101–109. [CrossRef]
- Ren, X.; Guo, H.; Li, S.; Wang, S.; Li, J. A novel image classification method with CNN-XGBoost model. In Proceedings of the Digital Forensics and Watermarking: 16th International Workshop, IWDW 2017, Magdeburg, Germany, 2017, Proceedings 16. Springer, 2017, August 23-25; pp. 378–390. [CrossRef]
Figure 1.
Graphic representation of the CRISPR cluster. The Cas cluster comprises genes coding for the needed proteins for the correct function of the system; the leader sequence controls the expression of the CRISPR array when it becomes necessary; and repeats are strongly conserved palindromic sequences capable of hairpin formation.
Figure 1.
Graphic representation of the CRISPR cluster. The Cas cluster comprises genes coding for the needed proteins for the correct function of the system; the leader sequence controls the expression of the CRISPR array when it becomes necessary; and repeats are strongly conserved palindromic sequences capable of hairpin formation.

Figure 2.
Stages for immunity acquisition after infection. (1) The Cas1-Cas2 multimeric complex recognizes the invading viral DNA, from which a sequence known as the "protospacer" is cut. (2) Sequentially, the complex integrates the protospacer upstream of the CRISPR array, exactly next to the leader sequence. (3) When the bacteriophage reinfects, the leader sequence initiates the CRISPR array expression, which yields the pre-crRNA. Then, it undergoes selective ribonucleotide sequence degradation by RNase III with (4) parallel binding of the tracrRNA to the desired sequence, generating the crRNA:tracrRNA complex. (5) The latter complex binds to the Cas9 protein, which digs into the viral DNA to target the complementary sequence to the crRNA for its cleavage.
Figure 2.
Stages for immunity acquisition after infection. (1) The Cas1-Cas2 multimeric complex recognizes the invading viral DNA, from which a sequence known as the "protospacer" is cut. (2) Sequentially, the complex integrates the protospacer upstream of the CRISPR array, exactly next to the leader sequence. (3) When the bacteriophage reinfects, the leader sequence initiates the CRISPR array expression, which yields the pre-crRNA. Then, it undergoes selective ribonucleotide sequence degradation by RNase III with (4) parallel binding of the tracrRNA to the desired sequence, generating the crRNA:tracrRNA complex. (5) The latter complex binds to the Cas9 protein, which digs into the viral DNA to target the complementary sequence to the crRNA for its cleavage.

Figure 3.
Single guide RNA (sgRNA). Rather than possessing a double RNA of crRNA and tracrRNA, a single RNA is synthesized using a linker loop, creating the single guide RNA.
Figure 3.
Single guide RNA (sgRNA). Rather than possessing a double RNA of crRNA and tracrRNA, a single RNA is synthesized using a linker loop, creating the single guide RNA.

Figure 4.
RNA sequence prediction using machine learning algorithms. (1) Random Forest is an ensemble learning method that uses a combination of decision trees to classify RNA sequences. It creates multiple decision trees on different subsets of the dataset and combines their predictions to obtain the final output. Decision trees work the same as Random Forest but with only one branch. (2) Support Vector Machine (SVM) is a supervised learning method that separates RNA sequences into two classes. SVM is known for its ability to handle non-linear data.
Figure 4.
RNA sequence prediction using machine learning algorithms. (1) Random Forest is an ensemble learning method that uses a combination of decision trees to classify RNA sequences. It creates multiple decision trees on different subsets of the dataset and combines their predictions to obtain the final output. Decision trees work the same as Random Forest but with only one branch. (2) Support Vector Machine (SVM) is a supervised learning method that separates RNA sequences into two classes. SVM is known for its ability to handle non-linear data.
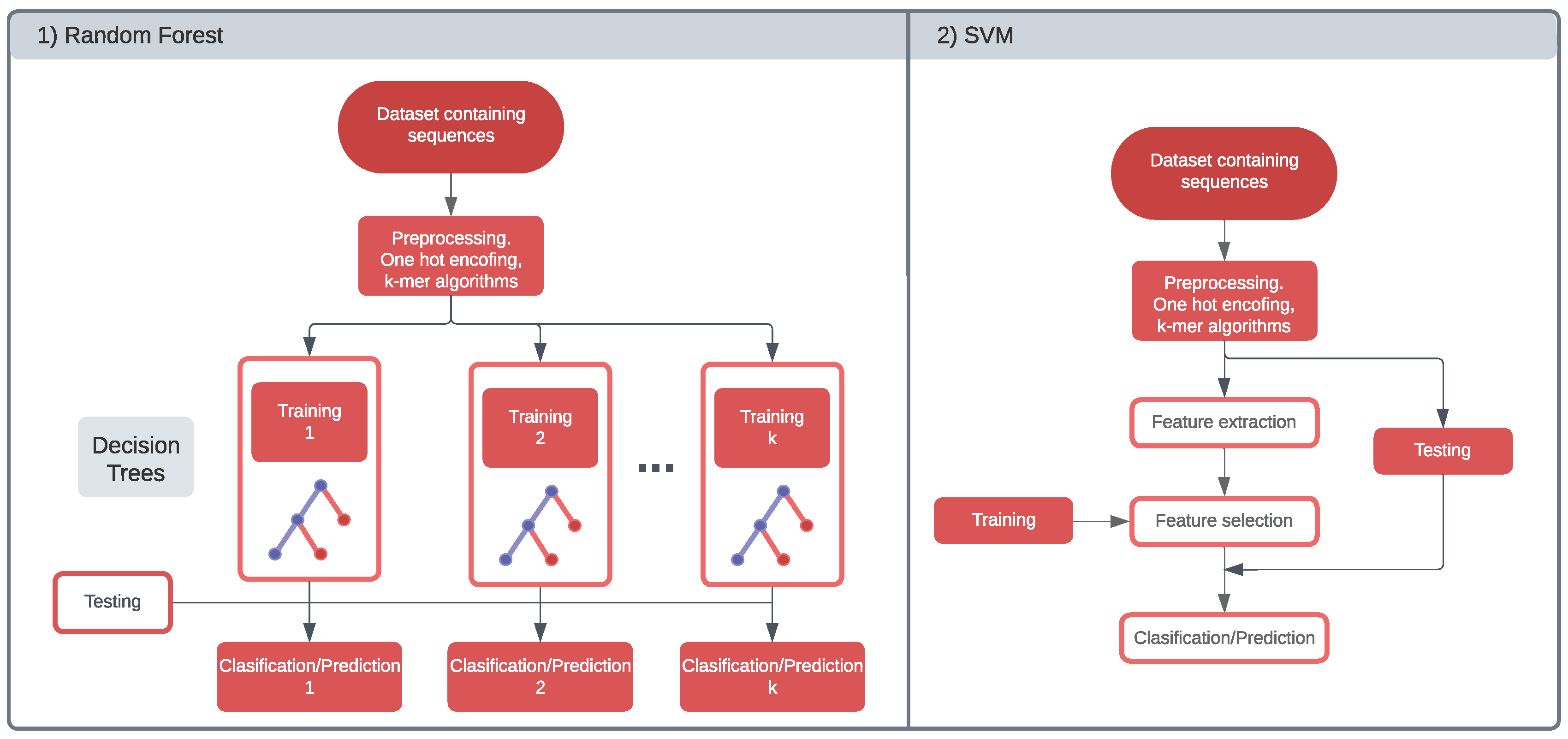
Figure 5.
CRISPR prediction using machine learning algorithms. (1) It takes RNA sequences as input and processes them through multiple layers of recurrent neurons. The output of each layer is fed back as input to the next layer, allowing the network to capture the temporal dependencies between nucleotides. The final output is a predicted RNA sequence. (2) It uses a series of convolutional layers to extract features from the RNA sequence, followed by a fully connected layer to make the prediction. The convolutional layers apply filters to the input sequence to detect specific patterns, such as base pairs or motifs, and the fully connected layer combines these patterns to generate the predicted RNA sequence
Figure 5.
CRISPR prediction using machine learning algorithms. (1) It takes RNA sequences as input and processes them through multiple layers of recurrent neurons. The output of each layer is fed back as input to the next layer, allowing the network to capture the temporal dependencies between nucleotides. The final output is a predicted RNA sequence. (2) It uses a series of convolutional layers to extract features from the RNA sequence, followed by a fully connected layer to make the prediction. The convolutional layers apply filters to the input sequence to detect specific patterns, such as base pairs or motifs, and the fully connected layer combines these patterns to generate the predicted RNA sequence

Figure 6.
Timeline from 2013 to 2017. Machine learning-based tools filled the first years. Many of these tools utilized datasets published by other ones for the training stage. Other tools noted their weaknesses, and time later launched their respective upgrades, dealing with the identified problems.
Figure 6.
Timeline from 2013 to 2017. Machine learning-based tools filled the first years. Many of these tools utilized datasets published by other ones for the training stage. Other tools noted their weaknesses, and time later launched their respective upgrades, dealing with the identified problems.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.


Docker4Circ: A Framework for a Reproducible Characterization of CircRNAs from RNA-Seq Data
Giulio Ferrero
et al.
,
2019
CRISPR-Cas Systems in Gut Microbiome of Children with Autism Spectrum Disorders
Natalia V. Zakharevich
et al.
,
2022
Cloning, Expression, and Functional Analysis of the Compact Anoxybacillus flavithermus Cas9 Nuclease
Anastasiya Matveeva
et al.
,
2023



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated