
Preprint
Article
A Two-Sample Mendelian Randomization Analysis Investigates Associations Between Gut Microbiota and Dyslipidemia
Altmetrics
Downloads
125
Views
42
Comments
0
A peer-reviewed article of this preprint also exists.
supplementary.zip (2.53MB )


This version is not peer-reviewed
Submitted:
15 September 2023
Posted:
18 September 2023
You are already at the latest version
Alerts
Abstract
The determination of a causal relationship between gut microbiota and a range of dyslipidemia remains uncertain. To clarify these associations, we employed a two-sample mendelian randomization (MR) analysis utilizing the inverse-variance weighted (IVW) method. This comprehensive analysis investigated the genetic variants that exhibited a significant association (p<1e-5) with 129 distinct gut microbiota genera, and their potential link to diverse forms of dyslipidemia. The results indicated a potential causal relationship between 22 gut microbiota genera and dyslipidemia in humans. Furthermore, these findings suggested that the impact of gut microbiota on dyslipidemia regulation is dependent on the specific phylum, family, and genus. Bacillota phylum demonstrated the greatest diversity, with 15 distinct genera distributed among 8 families. Notably, gut microbiota derived from the Lachnospiraceae and Lactobacillaceae families exhibit statistically significant associations with lipid levels that contribute to overall health (p<0.05). The sensitivity analysis indicated that our findings possess robustness (p>0.05). The findings of our investigation provide compelling evidence that supports a causal relationship between the gut microbiota and dyslipidemia in the human body. It is noteworthy to highlight the significant influence of the Bacillota phylum as a pivotal regulator of lipid levels, and the families Lachnospiraceae and Lactobacillaceae should be acknowledged as probiotics that make substantial contributions to this metabolic process.
Keywords:
Subject: Medicine and Pharmacology - Dietetics and Nutrition
1. Introduction
The gut microecosystem, consisting of approximately 1,014 microorganisms [1], is the most extensive, intricate, and vulnerable microecosystem within the human body[2]. It assumes a crucial role in both human health and diseases. Among the various microorganisms present in this microecosystem, the gut microbiota, including bacteria, viruses, fungi, and other microorganisms, is a substantial constituent[3], with bacteria accounting for more than 95% of the overall population[4]. The significance of gut microbiota has been increasingly validated through extensive research. Firstly, the establishment of normal intestinal flora through enteral colonization is imperative for the maintenance of intestinal barrier function[5]. Secondly, gut microbiota bestow various advantages on the host, including intestinal, immune, and nutritional benefits[6], thereby facilitating digestion, and regulating gut hormone secretion and physiological development, and defensing against pathogen colonization[7,8,9]. Altered gut microbiota is currently believed to have a significant impact on not only intestinal disorders but also a range of disease conditions[10]. Recent studies have demonstrated a close association between changes in gut microbiota and various health issues, including diabetes[11,12], obesity[13,14,15,16], chronic kidney disease (CKD)[17,18,19], hyperlipidemia[20], cardiovascular disease[21,22], metabolic disturbances[23], colon cancer[24,25], and other intestinal diseases[26,27]. Furthermore, the investigation into the regulation of brain and behavior by gut microbiota encompasses various facets, including the intestinal nervous system[28], neuroimaging[29], the interplay between gut microbiota and the hostt[30,31,32,33], and the gut microbiota-intestinal-brain axis[34,35,36,37]. Moreover, individuals can employ flora transplantation to rectify disruptions in the host’s gut microbiota, thereby reinstating its normal and stable state and preserving the host’s intestinal equilibrium[38]. In conclusion, the intercommunication signals between the host and gut microbiota, encompassing the modulation of host metabolism by the gut microbiota, have the potential to impact the physiological well-being and pathological conditions of the host[39]. Extant literature has documented the prospective regulatory significance of the gut microbiota in lipid metabolism disorders[40], thereby proposing that manipulating the gut microbiota could serve as a pivotal approach in managing hyperlipidemias[41]. Furthermore, several studies have documented that the regulation of gut microbiota disorder, coupled with the inhibition of abnormal lipid metabolism, holds promise for ameliorating the advancement of liver injury[42]. These findings lend support to the potential impact of gut microbiota on lipid metabolism. Nevertheless, the causal association between gut microbiota and host lipid metabolism disorders remains inconclusive.
Dyslipidemia is presently characterized in clinical settings by the presence of anomalies in various lipid types, including high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), triglyceride (TG), total cholesterol (TC), apolipoprotein A1 (APOA1), and apolipoprotein B (APOB) concentrations[43,44,45,46]. Dyslipidemia can be regarded as a manifestation of lipid metabolism disorders or as a concomitant symptom of multiple diseases, including obesity[47], type 2 diabetes (T2D)[48], CKD[49,50,51], atherosclerosis and coronary heart diseas (CHD)[52,53,54], and malignant tumor[55,56,57,58]. It is widely recognized that elevated TG levels serve as not only a risk factor for acute pancreatitis[46] but also an independent “risk-enhancing factor” for atherosclerotic cardiovascular disease (ASCVD)[43,59]. In the case of patients with high or extremely high-risk ASCVD, prevailing guidelines emphasize the necessity of reducing low-density lipoprotein cholesterol (LDL-C) levels to the greatest extent possible in order to mitigate the occurrence of severe complications[60]. The levels of APOB protein have been found to have a positive correlation with hypercholesterolemia, and a decrease in APOB synthesis has been shown to significantly reduce LDL-C levels and the incidence of atherosclerosis[61,62]. Conversely, high levels of HDL-C have not been firmly established as a risk factor for CHD[63]. APOA1, a crucial component of HDL-C, contributes to over 70% of lipoproteins[64,65,66,67,68], which are also part of the HDL-C family and share a similar physiological function. These aforementioned pieces of evidence demonstrate the direct impact of lipid levels on the cardiovascular system. Due to the prevalence and significant impact of lipid abnormalities on overall health, this study aims to investigate the potential causal relationship between intestinal flora and lipid metabolism regulation in order to identify evidence supporting the use of intestinal flora modulation as a means of controlling lipid metabolic disorders. This research endeavor seeks to offer novel perspectives and ideas in this field.
Mendelian randomization (MR) analysis is a prevalent approach employed in population studies to evaluate causality, wherein genetic variation is utilized to ascertain the coherence between observed associations linking risk factors and outcomes[69,70]. The selection of genetic variation as an instrumental variable (IV) was employed in the implementation of Mendelian randomization (MR) to establish causality due to the random allocation and lifelong exposure of genetic alleles, thereby mitigating potential confounding factors inherent in the genetic process[71]. Furthermore, the majority of genetic variants frequently lack association with conventional epidemiological risk factors, rendering traditional epidemiological analysis techniques insufficient in accurately elucidating a causal relationship between genetic variants and diseases[72]. Mendelian randomization offers valuable guidance for investigations reliant on genetic variation, thereby mitigating or circumventing the bias induced by confounding factors inherent in traditional epidemiological methods[73,74,75]. In this current study, a MR analysis was conducted on a substantial community sample of European participants to investigate the causal association between various genus-based gut microbiota and dyslipidemia. By employing human genetic data within the MR framework, this study elucidates the impact of distinct gut microbiota genus on different types of dyslipidemia, thereby offering novel perspectives on the potential causal link between gut microbiota and dyslipidemia.
2. Material and Methods
2.1. Exposure Data
Genetic variants that exhibit a robust association with distinct genera of gut microbiota were identified through a comprehensive genome-wide association study (GWAS) conducted on individuals of European descent, as documented in the OpenGWAS database[76,77]. The study’s methodology is visually depicted in Figure s1. We conducted an IV screening using the “TwoSampleMR” R package[74,78,79] to obtain independent IVs that affect lipoprotein levels in various gut microbiota data sets. The parameters used were as follows: p1 = 5e-8 (genetic variants must exhibit a strong association with the exposure), clump=TRUE, r2=0.01, kb=5000 (IVs with linkage disequilibrium were removed to ensure the independence of the selected genetic variations)[80,81]. A comprehensive screening process was conducted on 129 potential datasets to identify IVs for exposure, with their corresponding GWAS IDs ranging from “EBI-A-GCST90016959” to “EBI-A-GCST90017087” (Table S1).
2.2. Outcome Data
SNPs associated with dyslipidemia (HDL-C, LDL-C, TG, TC, APOA1, APOB) were also obtained from the OpenGWAS database, and the population structure is also dominated by European (Table S1). If there were two studies with overlapping data, the study with the largest sample size was included. In this step, we intersected the independent IVs from exposure factors and single nucleotide polymorphisms (SNPs) of outcome event and constructed a relationship of “independent exposure IV” - “factors” - “outcome variables” and eliminated SNPs associated with potential confounding variables via the PhenoScanner [82,83] database (http://www.phenos-canner.medschl.cam.ac.uk/phenoscanner). Then, we combined the two sets of data for subsequent MR analysis.
2.3. Ethics Statement
The present study utilized publicly accessible GWAS summary statistics data sourced from the OpenGWAS database. This database obtained informed consent from all participating studies in accordance with the protocols approved by their respective institutional review boards. Consequently, the submission of a dedicated ethics statement is unnecessary.
2.4. Statistical Analysis
The standard inverse-variance weighted (IVW) method was employed for primary two-sample Mendelian randomization (MR) analyses, which were further enhanced by incorporating the weighted median and MR Egger methods available in the TwoSampleMR package[78,84]. The study aimed to examine the variability in the relationship between different genus of gut microbiota and different type of dislipidemia by utilizing Cochran’s Q statistics[48,85]. Heterogeneity was ascertained by assessing the significance of the p-value (less than 0.05) derived from the Q statistic. In cases where heterogeneity was present, the effect evaluation was estimated using the random-effects IVW method, while the fixed-effects model was employed in the absence of heterogeneity[86,87]. Sensitivity analyses were conducted to identify and address potential pleiotropy in the causal estimates[88,89,90]. Specifically, we assessed the presence of horizontal pleiotropy through MR-Egger regression, considering its intercept terms and the Mendelian randomization pleiotropy residual sum[78,91].When the intercept of the MR-Egger model deviates significantly from zero or its p value is less than 0.05, it suggests the presence of horizontal pleiotropy. In such cases, an alternative MR method was employed to report the findings[69,92]. For determining the final results, causal associations were considered statistically significant if the p value was less than 0.05.
3. Results
3.1. Dyslipidemia MR Estimates
In the context of two-sample MR Analysis, we have effectively discerned 6 gut microbiota genera that exhibit causality towards HDL-C (Figure 1A), 5 towards LDL-C (Figure 1B), 4 towards TC (Figure 1C), 4 towards TG (Figure 1D), 6 towards APOA1 (Figure 1E), and 6 towards APOB (Figure 1F). It is worth noting that the number of independent IVs employed varied across the different sets of causal relationships under investigation. Based on the final results, it is evident that the distribution of gut microbiota genera exhibiting a negative causal relationship with various forms of dislipidemia (OR<1, p value of IVW<0.05) can be outlined as follows: Coprobacter and Olsenella for HDL-C (Figure 1A), Peptococcus and Slackia for LDL-C (Figure 1B), Butyricicoccus and Enterorhabdus for TC (Figure 1C), Dorea and Ruminococcus torques group for TG (Figure 1D), Anaerotruncus, Coprobacter, and Ruminococcaceae UCG009 for APOA1 (Figure 1E), and Methanobrevibacter, Oscillospira, Peptococcus, and Ruminococcaceae UCG010 for APOB (Figure 1F). This observation suggests that an increase in the abundance of these bacterial genera in the gut is associated with a decrease in the production of the corresponding lipids. In contrast, the distribution of gut microbiota genera that exhibit a positive causal relationship with various types of dyslipidemia (OR>1, p value of IVW<0.05) is as follows: Coprococcus2, Lachnospiraceae NK4A136 group, Lactobacillus, and Parabacteroides for HDL-C (Figure 1A), Parasutterella, Ruminococcus2, and Terrisporobacter for LDL-C (Figure 1B), Eubacterium coprostanoligenes group and Lactococcus for TC (Figure 1C), Coprobacter and Olsenella for TG (Figure 1D), Lactobacillus, Parabacteroides, and Ruminococcaceae UCG010 for APOA1 (Figure 1E), and Parasutterella and Terrisporobacter for APOB (Figure 1F). These findings suggest that an increase in the abundance of these bacterial species in the gut is associated with elevated levels of the corresponding lipids. Supplementary Materials 1 provides comprehensive information regarding the association between statistically significant gut microbiota genera and various types of lipid disorders in the MR analysis.
3.2. Sensitivity Analyses
Sensitivity tests were conducted using the MR Egger test to examine the presence of horizontal pleiotropy in various gut microbiota genera with different types of dyslipidemia. The results indicated no significant evidence of horizontal pleiotropy, as indicated by p values greater than 0.05 for the MR-Egger regression intercept approach (Table s2). However, there is significant heterogeneity (p<0.05) in the causality between Olsenella and TG, Anaerotruncus, Ruminococcaceae UCG009 and APOA1, and the effect size for these relationship was estimated using the random effect model of the IVW method, while a fixed effects model was employed to assess other causal effect sizes. The ultimate findings demonstrated that all effect values were statistically significant (p<0.05, Table s2), thereby confirming the causal association between these gut microbiota genera and the regulation of lipid metabolism. Furthermore, the sensitivity analysis was performed using the leave-one-out method to assess the impact of individual SNPs on outcome estimation, and the findings consistently persisted (Supplementary materials 2). The scatter plot, depicting the MR estimate of the effect of various gut microbiota genera on different dyslipidemia types, exhibited a clear linear trend (Figure 2). The funnel plot demonstrated minimal heterogeneity (Supplementary materials 3). Collectively, these pieces of evidence strongly support the statistical robustness of the analysis results and the reliability of the conclusion.
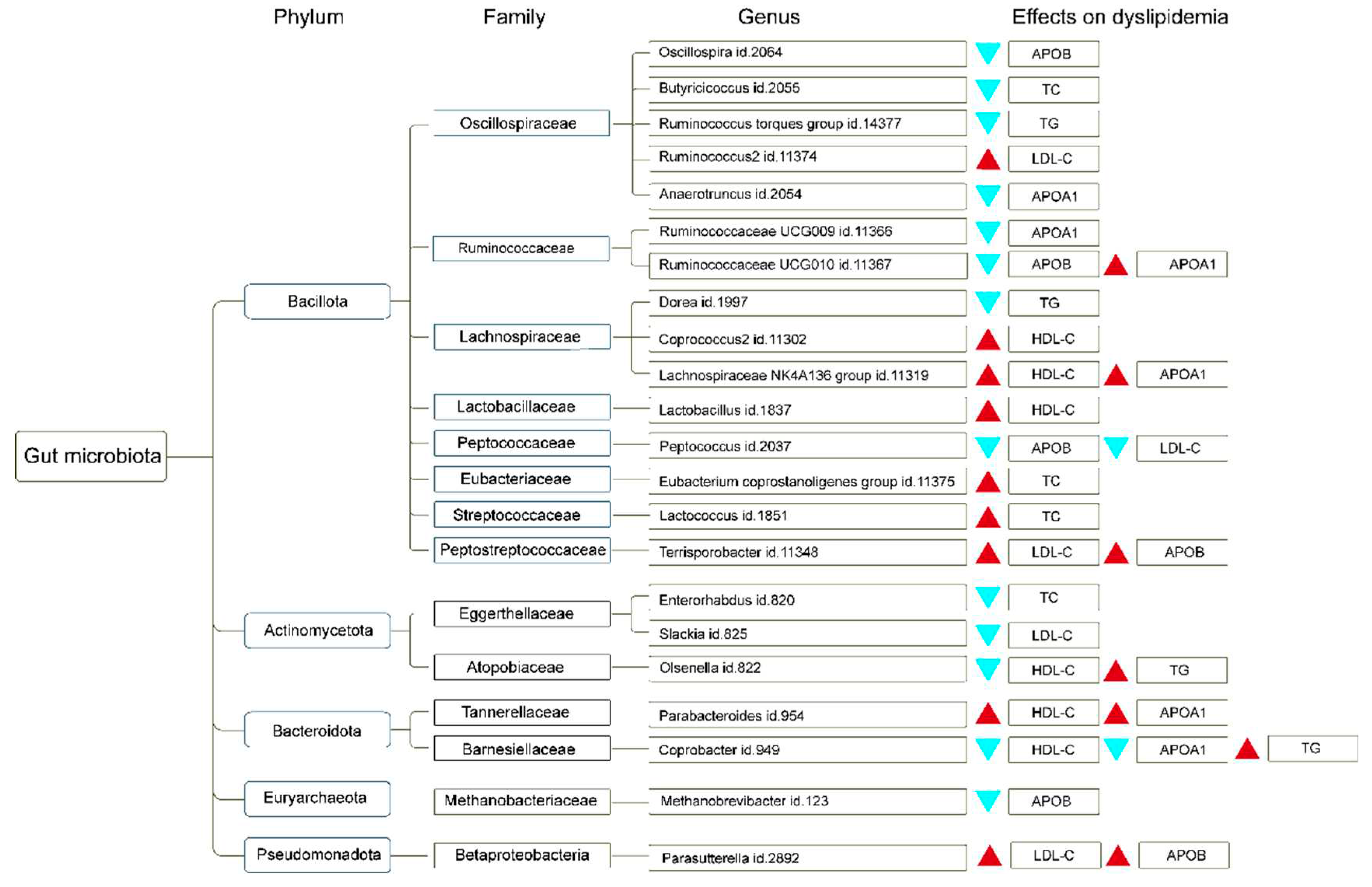
To enhance the understanding of the regulatory influence of gut microbiota genera on dyslipidemia, we summarized the phylum and family corresponding to different gut microbiota genera and their effects on different types of dyslipidemia, as shown in Figure 3.
4. Discussion
Dyslipidemia is a prominent manifestation of metabolic disorders and has emerged as a significant global public health concern, posing a substantial threat to human well-being[93,94,95]. Nonetheless, the etiology of dyslipidemia remains intricate and inconclusive. The gut microbiota, being the largest microbiota within the human body[6,96], assumes a crucial function in various aspects such as nutrition metabolism, growth and development, immunity, and the onset of diseases[16,97,98,99]. Despite the existing literature substantiating the association between gut microbiota and dyslipidemia[97], the presence of a causal link remains uncertain. To address this gap in knowledge, we employed MR analysis to investigate the potential causal relationships between various gut microbiota genera implicated in the regulation of lipid metabolism. Our findings yielded enlightening evidence in this regard. The findings of this investigation primarily highlight two pivotal observations: Firstly, the two-sample MR analysis has revealed a distinct causal relationship between gut microbiota and dyslipidemia, thereby presenting novel evidence regarding the involvement of gut microbiota in the regulation of physiological processes. Secondly, the inconsistent effects of gut microbiota originating from various taxonomic ranks, including different phylum, family, and genus, on lipid metabolism further substantiate the widespread and comprehensive influence of gut microbiota on the regulation of bodily functions. Overall, these findings will provide valuable insights for enhancing our understanding of the influence of gut microbiota on the physiological aspects of growth, development, and pathological conditions in the human body.
According to the observed distribution characteristics of bacterial phyla and families, our findings revealed the presence of up to 15 gut microbiota belonging to the Bacillota phylum and distributed across 8 distinct families, which also exhibited the highest phylum distribution among the gut microbiota identified in our study. Among them, we observed the presence of 5 distinct types of gut microbiota (Oscillospira, Butyricicoccus, Ruminococcus torques group, Ruminococcus2, and Anaerotruncus genus ) from the Oscillospiraceae family, each played distinct roles in lipid regulation. Ruminococcus2 and Anaerotruncus had the potential to increase lipid levels in the body, whereas other bacteria, such as Oscillospira, Butyricicoccus, and the Ruminococcus torques group genus, demonstrated the ability to decrease lipid levels. Oscillospiraceae is a bacterial family classified within the phylum Bacillota, consisting of obligate anaerobes. Despite the variation in shapes among its members, including rod-shaped and cocci forms[100], Oscillospira genus was recognized as a crucial types within the gut microbiota. Numerous studies have demonstrated a notable positive correlation between Oscillospira and low fat, leanness, constipation, and overall human health[101,102]. However, it is important to note that this organism has yet to be successfully cultured in isolation, and its metabolic and biological characteristics remain largely unknown[103]. In the present study, we have identified a negative regulatory association between Oscillospira and APOB levels, aligning with prior research on the physiological mechanisms by which Oscillospira modulate bodily functions, such as lower body mass index (BMI)[102]. These cumulative findings further augmented the plausibility of Oscillospira as prospective contenders for forthcoming probiotic interventions.
Based on available reports, the decrease in Butyricicoccus exhibited a strong correlation with inflammatory bowel disease (IBD)[104]. Moreover, emerging evidence indicates that IBD represents a collection of idiopathic inflammatory ailments distinguished by impaired intestinal immune system functionality and metabolic irregularities[105], and sphingolipid metabolism played a contributory role in the advancement of IBD[106]. In our study, we had discovered the significance of Butyricicoccus in the reduction of TC levels. This finding highlighted the potential regulatory function of Butyricicoccus in the body’s lipid metabolism and its association with disease processes related to lipid metabolism. Furthermore, this evidence contributed to our current understanding of the role of gut microbiota in the development of such diseases through the modulation of lipid metabolism. In our study, we identified two genera belonging to the Oscillospiraceae family[107], namely Ruminococcus torques group and Ruminococcus2. These genera exhibited distinct effects on dyslipidemia, with Ruminococcus torques group reducing lipid levels and Ruminococcus2 evaluating lipid levels. Previous research has reported a lower abundance of the Ruminococcus genus in individuals with IBD[108], Parkinson’s disease[109], or Amyotrophic lateral sclerosis[110,111]. Furthermore, Ruminococcus gnavus has been associated with Crohn’s disease[112].
In relation to the Ruminococcaceae family, we have identified two gut microbiota genera that exhibit distinct effects on lipid levels in the body. Specifically, the Ruminococcaceae UCG009 genus appears to decrease APOA1 levels, while the Ruminococcaceae UCG010 genus appears to reduce APOB levels. The Ruminococcaceae family is known to play a role in energy metabolism, insulin signaling, and inflammatory processes. Moreover, an increase in the relative abundance of Ruminococcaceae has been found to increase the risk of gestational diabetes mellitus (GDM) development[113]. In a study utilizing mice as a model, authors observed that Ruminococcaceae family exhibits a mitigating effect on fibrosis of non-alcoholic fatty liver disease (NAFLD)[114], and modulates hepatic fat content and lipid species composition[115].
The genera of gut microbiota belonging to the families Lachnospiraceae, Lactobacillaceae, and Peptococcaceae within the Bacillota phylum have demonstrated significant beneficial effects on lipid levels in the human body. Notably, the genera Dorea, Coprococcus2, and Lachnospiraceae NK4A136 group from the Lachnospiraceae family, Lactobacillus from the Lactobacillaceae family, and Peptococcus from the Peptococcaceae family have exhibited a dual role in lipid metabolism regulation. These gut microbiota have the ability to reduce harmful lipids (APOB and LDL-C) while also promoting the evaluation of beneficial lipids (HDL-C and APOA1) in the body. Lachnospiraceae, a prominent taxon in the human gut microbiota, has been found to potentially mitigate colon cancer in humans through the production of butyric acid[116,117,118]. Additionally, it was reported that the reduction of Lachnospiraceae abundance has been associated with Chronic Spontaneous Urticaria[119], sleep deprivation[120], and obesity[121]. As is known to all, Lactobacillus genus plays a significant role in the microbiota of both humans and animals, particularly in various body sites such as the digestive and female genital systems[122]. Lactobacillus demonstrates a mutualistic symbiosis with the human body, wherein it serves to safeguard the host against potential pathogenic incursions, while the host reciprocally offers a nutrient source[123,124]. A randomized controlled trial (RCT) has discerned that Lactobacillus exerts a positive influence on glucose metabolism in pregnant women who are overweight or obese[125]. Our research findings indicate that Lactobacillus confer benefits in ameliorating aberrant lipid metabolism levels, aligning with previous investigations. These pieces of evidence contributed to the growing body of knowledge that underscores the involvement of Lactobacillus, as pivotal probiotics, in the physiological processes of the human body. The Peptococcus genus is classified as a Gram-positive bacterium genus within the family Peptococcaceae. Species belonging to this genus are commonly found in the human microbiome, particularly in the bacteria that constitute the gut flora. They are also present in the oral cavity, upper respiratory tract, and large intestine. Our findings further support a significant association between the Peptococcus genus and the reduction of LDL-C and APOB levels in the body, suggesting a potential role in the improvement of dyslipidemia.
Furthermore, our investigation revealed that various families belonging to the Bacillota phylum, including the Eubacterium coprostanoligenes group from the Eubacteriaceae family, Lactococcus from the Streptococcaceae family, and Terrisporobacter from the Peptostreptococcaceae family, play a significant role in enhancing lipid levels within the human body. Notably, these gut microbiota species exhibited pronounced impacts on TC and LDL-C levels. The significance of this family lies in the production of various strains that yield short chain fatty acids, notably butyric acid. These short chain fatty acids are widely acknowledged for their pivotal functions in upholding human well-being, encompassing their role as specialized nutrients and energy constituents of the intestinal epithelium, safeguarding the integrity of the intestinal mucosal barrier, mitigating inflammation levels in humans, and augmenting gastrointestinal motility[126,127]. Lactococcus, a beneficial microbiota, is frequently employed in the dairy industry for the production of fermented dairy products, including cheeses. However, our study has substantiated a positive causal association between Lactococcus and TC levels, thereby implying that individuals with elevated blood lipid levels should refrain from consuming cheese products. Terriporobacter, belonging to the Peptostrectococcaceae family, is presently under investigation to ascertain its distinctive attributes and biological mechanisms. Our research findings suggest that this particular gut microbiota exerts a heightened influence on LDL-C and APOB levels, thereby classifying it as a potentially beneficial or detrimental microbiota.
In addition, our findings reveal the presence of additional phyla in the observed data, including 3 gut microbiota belonging to the Actinomycetota phylum, which are distributed among 2families, 2 gut microbiota belonging to the Bacteroidota phylum are distributed across 2 families. Moreover, within the Euryarchaeota and Pseudomonadota phylum, 2 distinct gut microbiota genera are identified, each belonging to their respective autonomous families. The Actinomycetota genus is prevalent in the microbiome of human infants[128] and is known for its production of bioactive metabolites with medicinal value[129]. Our study reveals a robust causal relationship between Eggerthellaceae and the reduction of TC and LDL-C levels in the human body. Conversely, the presence of Atopobiaceae bacteria is associated with elevated blood lipid levels, resulting in increased TG levels and decreased HDL-C levels. In a similar vein, two distinct families of gut microbiota, Tannellaceae and Barnesiellaceae, affiliated with the Bacteroidota phylum, have exhibited inconsistent impacts on the regulation of lipid metabolism. Specifically, Tannellaceae bacteria have demonstrated the capacity to augment levels of HDL-C and APOA1, thereby potentially mitigating the rise of blood lipid levels. Conversely, the presence of Barnesiellaceae has been observed to engender a reduction in HDL-C and APOA1 levels, concomitant with an elevation in TG levels. The Metanobacteriaceae family, which falls under the Euryarchaeota phylum, has been recognized as a pathogenic microorganism. Our investigation reveals that this particular gut microbiota exerts a suppressive impact on APOB levels. Furthermore, the existence of the Betaproteobacteria family from the Pseudomonas phylum demonstrates a significant positive causal relationship with increased levels of LDL-C and APOB. This implies a distinct inclination of this bacterial family to stimulate elevated lipid levels within the human body.
In conclusion, the influence of gut microbiota on lipid metabolism varies depending on the specific types of gut microbiota. Our study demonstrates that the predominant phylum of gut microbiota in humans also encompasses the most diverse microbial group responsible for regulating lipid metabolism. Notably, Lachnospiraceae and Lactobacillaceae families play a significant role and should be recognized as a key microbiota in ameliorating lipid metabolism abnormalities within the body. Furthermore, it is imperative to acknowledge that individuals with concomitant hyperlipidemia should refrain from consuming cheese. Our research findings elucidate the wide-ranging and ubiquitous influence of gut microbiota on the regulation of lipid metabolism levels, thereby enhancing our comprehension of the interplay between gut microbiota and diseases associated with lipid disorders. These results provide novel evidence that contributes to a more comprehensive understanding of how gut microbiota modulates bodily functions and metabolism.
Our study possesses several notable strengths, such as the implementation of the MR approach, which effectively mitigates certain confounding factors frequently encountered in epidemiological studies. Additionally, we have employed a homogenous population, thereby reducing the inherent heterogeneity often encountered when individuals from diverse ancestral backgrounds are included in genetic studies. Stratified analyses were employed to assess the causal relationships between various genus of gut microbiota and distinct dyslipidemia types. Additionally, sensitivity analysis was conducted on the subgroup analysis outcomes, yielding statistically robust results. Nevertheless, it is important to note that the inclusion of exclusively European individuals in our analyses may limit the generalizability of these findings to other ancestral populations.
5. Conclusions
Our findings demonstrate a definitive causal link between gut microbiota and lipid abnormalities in the human body. Specifically, the Bacillota phylum exhibits the most extensive regulation of body lipid levels. The families Lachnospiraceae and Lactobacillaceae play a significant role in ameliorating lipid metabolism abnormalities and should be recognized as key microbiota in this process. Additionally, it is recommended that individuals with hyperlipidemia refrain from cheese consumption due to the potential detrimental impact of lactococcus, which is abundant in cheese, on their lipid profiles.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
MJ.Z and ZJ.F designed the study. XY.Z and PQ.L analysed the data and drafted the paper. H.L and YH.W critically revised the paper. All authors read and approved the final manuscript.
Funding
This research was funded by Guangdong Basic and Applied Basic Research Foundation, grant number 2020A1515110543.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
All data used in this study are available in the public repository. The code involved in the data analysis process can be obtained by contacting the corresponding author.
Acknowledgments
We thank the team of OpenGWAS and UK Biobank database for making the summary data publicly available, and we would like to acknowledge the principal investigators of the studies who made their data openly accessible for research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kamada, N.; Seo, S.-U.; Chen, G.Y.; Núñez, G. Role of the gut microbiota in immunity and inflammatory disease. Nature Reviews Immunology 2013, 13, 321–335. [Google Scholar] [CrossRef]
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006, 124, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lian, J.; Zheng, Q.; Wang, L.; Wang, Y.; Yang, D. Composition analysis and prebiotics properties of polysaccharides extracted from Lepista sordida submerged cultivation mycelium. Frontiers in microbiology 2022, 13, 1077322. [Google Scholar] [CrossRef]
- Bi, C.R.; Sun, J.T.; Du, J.; Chu, L.Y.; Li, Y.J.; Jia, X.Y.; Liu, Y.; Zhang, W.P.; Li, Y.C.; Liu, Y.J. Effects of Zhishi Daozhi Decoction on the intestinal flora of nonalcoholic fatty liver disease mice induced by a high-fat diet. Frontiers in cellular and infection microbiology 2022, 12, 1005318. [Google Scholar] [CrossRef]
- Luo, Q.; Lei, X.; Xu, J.; Jahangir, A.; He, J.; Huang, C.; Liu, W.; Cheng, A.; Tang, L.; Geng, Y. , et al. An altered gut microbiota in duck-origin parvovirus infection on cherry valley ducklings is associated with mucosal barrier dysfunction. Poultry science 2021, 100, 101021. [Google Scholar] [CrossRef]
- Makrgeorgou, A.; Leonardi-Bee, J.; Bath-Hextall, F.J.; Murrell, D.F.; Tang, M.L.; Roberts, A.; Boyle, R.J. Probiotics for treating eczema. Cochrane Database Syst Rev 2018, 11, Cd006135. [Google Scholar] [CrossRef]
- Mishra, A.K.; Dubey, V.; Ghosh, A.R. Obesity: An overview of possible role(s) of gut hormones, lipid sensing and gut microbiota. Metabolism: clinical and experimental 2016, 65, 48–65. [Google Scholar] [CrossRef]
- Wei, M.Y.; Shi, S.; Liang, C.; Meng, Q.C.; Hua, J.; Zhang, Y.Y.; Liu, J.; Zhang, B.; Xu, J.; Yu, X.J. The microbiota and microbiome in pancreatic cancer: more influential than expected. Molecular cancer 2019, 18, 97. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat Med 2018, 24, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Vuotto, C.; Longo, F.; Donelli, G. Probiotics to counteract biofilm-associated infections: promising and conflicting data. International journal of oral science 2014, 6, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Wu, K.; Xu, L.; Cen, Y.; Ni, J.; Chen, J.; Zheng, W.; Liu, W. Methanol extract of Inonotus obliquus improves type 2 diabetes mellitus through modifying intestinal flora. Frontiers in endocrinology 2022, 13, 1103972. [Google Scholar] [CrossRef] [PubMed]
- Pasini, E.; Corsetti, G.; Assanelli, D.; Testa, C.; Romano, C.; Dioguardi, F.S.; Aquilani, R. Effects of chronic exercise on gut microbiota and intestinal barrier in human with type 2 diabetes. Minerva medica 2019, 110, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.B.; Jun, D.W.; Kang, B.K.; Lim, J.H.; Lim, S.; Chung, M.J. Randomized, Double-blind, Placebo-controlled Study of a Multispecies Probiotic Mixture in Nonalcoholic Fatty Liver Disease. Scientific reports 2019, 9, 5688. [Google Scholar] [CrossRef] [PubMed]
- Al-Assal, K.; Martinez, A.C.; Torrinhas, R.S.; Cardinelli, C.; Waitzberg, D. Gut microbiota and obesity. Clinical Nutrition Experimental 2018, 20, 60–64. [Google Scholar] [CrossRef]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, inflammation, and metabolic disease. Cell metabolism 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhao, Q.; Liu, J.; Huang, A.; Xia, X. Buyang Huanwu decoction affects gut microbiota and lipid metabolism in a ZDF rat model of co-morbid type 2 diabetes mellitus and obesity: An integrated metabolomics analysis. Frontiers in chemistry 2022, 10, 1036380. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Clément, K. The gut microbiome, diet, and links to cardiometabolic and chronic disorders. Nature reviews. Nephrology 2016, 12, 169–181. [Google Scholar] [CrossRef]
- Chung, S.; Barnes, J.L.; Astroth, K.S. Gastrointestinal Microbiota in Patients with Chronic Kidney Disease: A Systematic Review. Advances in nutrition (Bethesda, Md.) 2019, 10, 888–901. [Google Scholar] [CrossRef]
- Feng, Y.L.; Cao, G.; Chen, D.Q.; Vaziri, N.D.; Chen, L.; Zhang, J.; Wang, M.; Guo, Y.; Zhao, Y.Y. Microbiome-metabolomics reveals gut microbiota associated with glycine-conjugated metabolites and polyamine metabolism in chronic kidney disease. Cellular and molecular life sciences : CMLS 2019, 76, 4961–4978. [Google Scholar] [CrossRef]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B. , et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef]
- Jie, Z.; Xia, H.; Zhong, S.L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H. , et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat Commun 2017, 8, 845. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, C. Analysis of changes in intestinal flora and intravascular inflammation and coronary heart disease in obese patients. Experimental and therapeutic medicine 2018, 15, 4538–4542. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.; Carvalho, D.; Freitas, P. Gut Microbiota: Association with NAFLD and Metabolic Disturbances. BioMed research international 2015, 2015, 979515. [Google Scholar] [CrossRef] [PubMed]
- Rezasoltani, S.; Asadzadeh-Aghdaei, H.; Nazemalhosseini-Mojarad, E.; Dabiri, H.; Ghanbari, R.; Zali, M.R. Gut microbiota, epigenetic modification and colorectal cancer. Iranian journal of microbiology 2017, 9, 55–63. [Google Scholar] [PubMed]
- Wang, X.; Yang, Y.; Huycke, M.M. Commensal-infected macrophages induce dedifferentiation and reprogramming of epithelial cells during colorectal carcinogenesis. Oncotarget 2017, 8, 102176–102190. [Google Scholar] [CrossRef]
- Li, S.; Wang, Z.; Yang, Y.; Yang, S.; Yao, C.; Liu, K.; Cui, S.; Zou, Q.; Sun, H.; Guo, G. Lachnospiraceae shift in the microbial community of mice faecal sample effects on water immersion restraint stress. AMB Express 2017, 7, 82. [Google Scholar] [CrossRef]
- Armstrong, H.; Alipour, M.; Valcheva, R.; Bording-Jorgensen, M.; Jovel, J.; Zaidi, D.; Shah, P.; Lou, Y.; Ebeling, C.; Mason, A.L. , et al. Host immunoglobulin G selectively identifies pathobionts in pediatric inflammatory bowel diseases. Microbiome 2019, 7, 1. [Google Scholar] [CrossRef]
- Furness, J.B. The enteric nervous system and neurogastroenterology. Nature Reviews Gastroenterology & Hepatology 2012, 9, 286–294. [Google Scholar] [CrossRef]
- Mayer, E.A.; Aziz, Q.; Coen, S.; Kern, M.; Labus, J.S.; Lane, R.; Kuo, B.; Naliboff, B.; Tracey, I. Brain imaging approaches to the study of functional GI disorders: a Rome working team report. Neurogastroenterology and motility 2009, 21, 579–596. [Google Scholar] [CrossRef]
- Subramanya, S.H.; Sharan, N.K.; Baral, B.P.; Hamal, D.; Nayak, N.; Prakash, P.Y.; Sathian, B.; Bairy, I.; Gokhale, S. Diversity, in-vitro virulence traits and antifungal susceptibility pattern of gastrointestinal yeast flora of healthy poultry, Gallus gallus domesticus. BMC microbiology 2017, 17, 113. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nature reviews. Immunology 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Liu, Y.; Chen, J.; Bai, Y.; He, J.; Cao, H.; Che, Q.; Guo, J.; Su, Z. Marine Chitooligosaccharide Alters Intestinal Flora Structure and Regulates Hepatic Inflammatory Response to Influence Nonalcoholic Fatty Liver Disease. Marine drugs 2022, 20. [Google Scholar] [CrossRef] [PubMed]
- Bo, L.; Li, J.; Tao, T.; Bai, Y.; Ye, X.; Hotchkiss, R.S.; Kollef, M.H.; Crooks, N.H.; Deng, X. Probiotics for preventing ventilator-associated pneumonia. Cochrane Database Syst Rev 2014, 10, Cd009066. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.M.; Surette, M.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nature Reviews Microbiology 2012, 10, 735–742. [Google Scholar] [CrossRef]
- Bercik, P. The microbiota–gut–brain axis: learning from intestinal bacteria? 2011, 60, 288–289. [CrossRef]
- Collins, S.M.; Bercik, P. The relationship between intestinal microbiota and the central nervous system in normal gastrointestinal function and disease. Gastroenterology 2009, 136, 2003–2014. [Google Scholar] [CrossRef]
- Yi, X.; Zhou, K.; Deng, N.; Cai, Y.; Peng, X.; Tan, Z. Simo decoction curing spleen deficiency constipation was associated with brain-bacteria-gut axis by intestinal mucosal microbiota. Frontiers in microbiology 2023, 14, 1090302. [Google Scholar] [CrossRef]
- El-Salhy, M.; Winkel, R.; Casen, C.; Hausken, T.; Gilja, O.H.; Hatlebakk, J.G. Efficacy of Fecal Microbiota Transplantation for Patients With Irritable Bowel Syndrome at 3 Years After Transplantation. Gastroenterology 2022, 163, 982–994. [Google Scholar] [CrossRef]
- Kindt, A.; Liebisch, G.; Clavel, T.; Haller, D.; Hörmannsperger, G.; Yoon, H.; Kolmeder, D.; Sigruener, A.; Krautbauer, S.; Seeliger, C. , et al. The gut microbiota promotes hepatic fatty acid desaturation and elongation in mice. Nat Commun 2018, 9, 3760. [Google Scholar] [CrossRef]
- You, H.; Deng, X.; Bai, Y.; He, J.; Cao, H.; Che, Q.; Guo, J.; Su, Z. The Ameliorative Effect of COST on Diet-Induced Lipid Metabolism Disorders by Regulating Intestinal Microbiota. Marine drugs 2022, 20. [Google Scholar] [CrossRef]
- Qin, S.; He, Z.; Wu, Y.; Zeng, C.; Zheng, Z.; Zhang, H.; Lv, C.; Yuan, Y.; Wu, H.; Ye, J. , et al. Instant Dark Tea Alleviates Hyperlipidaemia in High-Fat Diet-Fed Rat: From Molecular Evidence to Redox Balance and Beyond. Frontiers in nutrition 2022, 9, 819980. [Google Scholar] [CrossRef]
- Liu, H.; Meng, W.; Zhao, D.; Ma, Z.; Zhang, W.; Chen, Z.; Li, Z.; Zhao, P. Study on mechanism of action of total flavonoids from Cortex Juglandis Mandshuricae against alcoholic liver disease based on “gut-liver axis”. Frontiers in pharmacology 2022, 13, 1074286. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.J.; Ginsberg, H.N.; Amarenco, P.; Andreotti, F.; Borén, J.; Catapano, A.L.; Descamps, O.S.; Fisher, E.; Kovanen, P.T.; Kuivenhoven, J.A. , et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. European heart journal 2011, 32, 1345–1361. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Hólm, H.; Ding, E.L.; Johnson, T. , et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet (London, England) 2012, 380, 572–580. [Google Scholar] [CrossRef]
- Holmes, M.V.; Asselbergs, F.W.; Palmer, T.M.; Drenos, F.; Lanktree, M.B.; Nelson, C.P.; Dale, C.E.; Padmanabhan, S.; Finan, C.; Swerdlow, D.I. , et al. Mendelian randomization of blood lipids for coronary heart disease. European heart journal 2015, 36, 539–550. [Google Scholar] [CrossRef] [PubMed]
- groups, E. Emergency expert consensus on diagnosis and treatment of hypertriglyceridemic acute pancreatitis. Chinese General Practice 2021, 24, 3781–3793. [Google Scholar]
- Klop, B.; Elte, J.W.; Cabezas, M.C. Dyslipidemia in obesity: mechanisms and potential targets. Nutrients 2013, 5, 1218–1240. [Google Scholar] [CrossRef]
- Soremekun, O.; Karhunen, V.; He, Y.; Rajasundaram, S.; Liu, B.; Gkatzionis, A.; Soremekun, C.; Udosen, B.; Musa, H.; Silva, S. , et al. Lipid traits and type 2 diabetes risk in African ancestry individuals: A Mendelian Randomization study. EBioMedicine 2022, 78, 103953. [Google Scholar] [CrossRef]
- Bulbul, M.C.; Dagel, T.; Afsar, B.; Ulusu, N.N.; Kuwabara, M.; Covic, A.; Kanbay, M. Disorders of Lipid Metabolism in Chronic Kidney Disease. Blood purification 2018, 46, 144–152. [Google Scholar] [CrossRef]
- Moradi, H.; Vaziri, N.D. Molecular mechanisms of disorders of lipid metabolism in chronic kidney disease. Frontiers in bioscience (Landmark edition) 2018, 23, 146–161. [Google Scholar] [CrossRef]
- Ferro, C.J.; Mark, P.B.; Kanbay, M.; Sarafidis, P.; Heine, G.H.; Rossignol, P.; Massy, Z.A.; Mallamaci, F.; Valdivielso, J.M.; Malyszko, J. , et al. Lipid management in patients with chronic kidney disease. Nature reviews. Nephrology 2018, 14, 727–749. [Google Scholar] [CrossRef]
- Ou, M.; Li, X.; Zhao, S.; Cui, S.; Tu, J. Long non-coding RNA CDKN2B-AS1 contributes to atherosclerotic plaque formation by forming RNA-DNA triplex in the CDKN2B promoter. EBioMedicine 2020, 55, 102694. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nature reviews. Disease primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Sheng, W.; Hu, G. To Analyze the Influencing Factors of Senile Coronary Heart Disease Patients Complicated with Frailty Syndrome. Journal of healthcare engineering 2022, 2022, 7619438. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zheng, L.; Xie, X.; Luo, J.; Yu, J.; Zhang, L.; Meng, W.; Zhou, Y.; Chen, L.; Ouyang, D. , et al. Targeting PLA2G16, a lipid metabolism gene, by Ginsenoside Compound K to suppress the malignant progression of colorectal cancer. Journal of advanced research 2022, 36, 265–276. [Google Scholar] [CrossRef]
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid metabolism and cancer. The Journal of experimental medicine 2021, 218. [Google Scholar] [CrossRef]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer communications (London, England) 2018, 38, 27. [Google Scholar] [CrossRef]
- Cao, Y. Adipocyte and lipid metabolism in cancer drug resistance. The Journal of clinical investigation 2019, 129, 3006–3017. [Google Scholar] [CrossRef]
- Virani, S.S.; Morris, P.B.; Agarwala, A.; Ballantyne, C.M.; Birtcher, K.K.; Kris-Etherton, P.M.; Ladden-Stirling, A.B.; Miller, M.; Orringer, C.E.; Stone, N.J. 2021 ACC Expert Consensus Decision Pathway on the Management of ASCVD Risk Reduction in Patients With Persistent Hypertriglyceridemia: A Report of the American College of Cardiology Solution Set Oversight Committee. Journal of the American College of Cardiology 2021, 78, 960–993. [Google Scholar] [CrossRef]
- Averna, M.; Banach, M.; Bruckert, E.; Drexel, H.; Farnier, M.; Gaita, D.; Magni, P.; März, W.; Masana, L.; Mello, E.S.A. , et al. Practical guidance for combination lipid-modifying therapy in high- and very-high-risk patients: A statement from a European Atherosclerosis Society Task Force. Atherosclerosis 2021, 325, 99–109. [Google Scholar] [CrossRef]
- Huang, Y.; Zheng, S.; Guo, Z.; de Mollerat du Jeu, X.; Liang, X.J.; Yang, Z.; Zhang, H.Y.; Gao, S.; Liang, Z. Ionizable liposomal siRNA therapeutics enables potent and persistent treatment of Hepatitis B. Signal transduction and targeted therapy 2022, 7, 38. [Google Scholar] [CrossRef]
- Richardson, T.G.; Sanderson, E.; Palmer, T.M.; Ala-Korpela, M.; Ference, B.A.; Davey Smith, G.; Holmes, M.V. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis. PLoS medicine 2020, 17, e1003062. [Google Scholar] [CrossRef] [PubMed]
- Boekholdt, S.M.; Arsenault, B.J.; Hovingh, G.K.; Mora, S.; Pedersen, T.R.; Larosa, J.C.; Welch, K.M.; Amarenco, P.; Demicco, D.A.; Tonkin, A.M. , et al. Levels and changes of HDL cholesterol and apolipoprotein A-I in relation to risk of cardiovascular events among statin-treated patients: a meta-analysis. Circulation 2013, 128, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Watts, G.F.; Coll, B.; Wasserman, S.M.; Marcovina, S.M.; Barrett, P.H.R. Lipoprotein(a) Particle Production as a Determinant of Plasma Lipoprotein(a) Concentration Across Varying Apolipoprotein(a) Isoform Sizes and Background Cholesterol-Lowering Therapy. Journal of the American Heart Association 2019, 8, e011781. [Google Scholar] [CrossRef] [PubMed]
- Di Angelantonio, E.; Sarwar, N.; Perry, P.; Kaptoge, S.; Ray, K.K.; Thompson, A.; Wood, A.M.; Lewington, S.; Sattar, N.; Packard, C.J. , et al. Major lipids, apolipoproteins, and risk of vascular disease. Jama 2009, 302, 1993–2000. [Google Scholar] [CrossRef]
- Sahm, A.; Bens, M.; Platzer, M.; Cellerino, A. Parallel evolution of genes controlling mitonuclear balance in short-lived annual fishes. Aging cell 2017, 16, 488–496. [Google Scholar] [CrossRef]
- Wilson, P.W.; Garrison, R.J.; Castelli, W.P.; Feinleib, M.; McNamara, P.M.; Kannel, W.B. Prevalence of coronary heart disease in the Framingham Offspring Study: role of lipoprotein cholesterols. The American journal of cardiology 1980, 46, 649–654. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, L.; Meng, L.; Liang, H.; Zhou, T.; Ye, S.; Qi, Z.; Huang, X.; Zhou, P.; Fu, W. Acupuncture treatment for carotid atherosclerotic plaques: study protocol for a pilot randomized, single blinded, controlled clinical trial. Trials 2020, 21, 768. [Google Scholar] [CrossRef]
- Emdin, C.A.; Khera, A.V.; Kathiresan, S. Mendelian Randomization. Jama 2017, 318, 1925–1926. [Google Scholar] [CrossRef]
- Birney, E. Mendelian Randomization. Cold Spring Harbor perspectives in medicine 2022, 12. [Google Scholar] [CrossRef]
- Lawlor, D.A.; Harbord, R.M.; Sterne, J.A.; Timpson, N.; Davey Smith, G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Statistics in medicine 2008, 27, 1133–1163. [Google Scholar] [CrossRef]
- Borges, M.C.; Oliveira, I.O.; Freitas, D.F.; Horta, B.L.; Ong, K.K.; Gigante, D.P.; Barros, A.J.D. Obesity-induced hypoadiponectinaemia: the opposite influences of central and peripheral fat compartments. International journal of epidemiology 2017, 46, 2044–2055. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Ebrahim, S. Mendelian randomization: prospects, potentials, and limitations. International journal of epidemiology 2004, 33, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Ebrahim, S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? International journal of epidemiology 2003, 32, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Riaz, H.; Khan, M.S.; Siddiqi, T.J.; Usman, M.S.; Shah, N.; Goyal, A.; Khan, S.S.; Mookadam, F.; Krasuski, R.A.; Ahmed, H. Association Between Obesity and Cardiovascular Outcomes: A Systematic Review and Meta-analysis of Mendelian Randomization Studies. JAMA network open 2018, 1, e183788. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.S.; Andrews, S.J.; Elsworth, B.; Gaunt, T.R.; Hemani, G.; Marcora, E. The variant call format provides efficient and robust storage of GWAS summary statistics. Genome Biol 2021, 22, 32. [Google Scholar] [CrossRef] [PubMed]
- Ben, E.; Matthew, L.; Tessa, A.; Yi, L.; Peter, M.; Jon, H.; Phil, B.; Tom, P.; Valeriia, H.; George Davey, S. , et al. The MRC IEU OpenGWAS data infrastructure. bioRxiv, 1101. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. International journal of epidemiology 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Rasooly, D.; Peloso, G.M. Two-Sample Multivariable Mendelian Randomization Analysis Using R. Current protocols 2021, 1, e335. [Google Scholar] [CrossRef]
- Li, Y.; Ma, L. Relationship between telomere length and the prognosis of breast cancer based on estrogen receptor status: A Mendelian randomization study. Frontiers in oncology 2022, 12, 1024772. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, M.; Chen, X.; Ruan, W.; Yao, J.; Lian, X. Telomere length and multiple sclerosis: a Mendelian randomization study. The International journal of neuroscience, 1080. [Google Scholar] [CrossRef]
- Kamat, M.A.; Blackshaw, J.A.; Young, R.; Surendran, P.; Burgess, S.; Danesh, J.; Butterworth, A.S.; Staley, J.R. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics (Oxford, England) 2019, 35, 4851–4853. [Google Scholar] [CrossRef]
- Staley, J.R.; Blackshaw, J.; Kamat, M.A.; Ellis, S.; Surendran, P.; Sun, B.B.; Paul, D.S.; Freitag, D.; Burgess, S.; Danesh, J. , et al. PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics (Oxford, England) 2016, 32, 3207–3209. [Google Scholar] [CrossRef]
- Davey Smith, G.; Hemani, G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Human molecular genetics 2014, 23, R89–R98. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; He, J.; Tian, F.F.; Bi, F.F.; Huang, K. A causal relationship between leukocyte telomere length and multiple sclerosis: A Mendelian randomization study. Frontiers in immunology 2022, 13, 922922. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Del Greco, M.F.; Minelli, C.; Davey Smith, G.; Sheehan, N.; Thompson, J. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Statistics in medicine 2017, 36, 1783–1802. [Google Scholar] [CrossRef] [PubMed]
- Kamiza, A.B.; Fatumo, S.; Singini, M.G.; Yeh, C.C.; Chikowore, T. Hepatitis B infection is causally associated with extrahepatic cancers: A Mendelian randomization study. EBioMedicine 2022, 79, 104003. [Google Scholar] [CrossRef]
- Yuan, S.; Carter, P.; Bruzelius, M.; Vithayathil, M.; Kar, S.; Mason, A.M.; Lin, A.; Burgess, S.; Larsson, S.C. Effects of tumour necrosis factor on cardiovascular disease and cancer: A two-sample Mendelian randomization study. EBioMedicine 2020, 59, 102956. [Google Scholar] [CrossRef]
- Yang, J.; He, X.; Qian, L.; Zhao, B.; Fan, Y.; Gao, F.; Yan, B.; Zhu, F.; Ma, X. Association between plasma proteome and childhood neurodevelopmental disorders: A two-sample Mendelian randomization analysis. EBioMedicine 2022, 78, 103948. [Google Scholar] [CrossRef]
- Ye, M.; Wang, Y.; Zhan, Y. Genetic association of leukocyte telomere length with Graves’ disease in Biobank Japan: A two-sample Mendelian randomization study. Frontiers in immunology 2022, 13, 998102. [Google Scholar] [CrossRef]
- Verbanck, M.; Chen, C.Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nature genetics 2018, 50, 693–698. [Google Scholar] [CrossRef]
- Zhan, Y.; Song, C.; Karlsson, R.; Tillander, A.; Reynolds, C.A.; Pedersen, N.L.; Hägg, S. Telomere Length Shortening and Alzheimer Disease--A Mendelian Randomization Study. JAMA neurology 2015, 72, 1202–1203. [Google Scholar] [CrossRef]
- Bourebaba, L.; Łyczko, J.; Alicka, M.; Bourebaba, N.; Szumny, A.; Fal, A.M.; Marycz, K. Inhibition of Protein-tyrosine Phosphatase PTP1B and LMPTP Promotes Palmitate/Oleate-challenged HepG2 Cell Survival by Reducing Lipoapoptosis, Improving Mitochondrial Dynamics and Mitigating Oxidative and Endoplasmic Reticulum Stress. Journal of clinical medicine 2020, 9. [Google Scholar] [CrossRef]
- Huang, P.L. A comprehensive definition for metabolic syndrome. Disease models & mechanisms 2009, 2, 231–237. [Google Scholar] [CrossRef]
- Qu, J.; Fourman, S.; Fitzgerald, M.; Liu, M.; Nair, S.; Oses-Prieto, J.; Burlingame, A.; Morris, J.H.; Davidson, W.S.; Tso, P. , et al. Low-density lipoprotein receptor-related protein 1 (LRP1) is a novel receptor for apolipoprotein A4 (APOA4) in adipose tissue. Scientific reports 2021, 11, 13289. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Yin, B.; Li, W.; Chai, T.; Liang, W.; Huang, Y.; Tan, X.; Zheng, P.; Wu, J.; Li, Y. , et al. Age-related changes in microbial composition and function in cynomolgus macaques. Aging 2019, 11, 12080–12096. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Zhao, N.; Zhang, L.; Chen, J.; Liu, X.; Piao, S. Gut microbiota is a potential goalkeeper of dyslipidemia. Frontiers in endocrinology 2022, 13, 950826. [Google Scholar] [CrossRef] [PubMed]
- Pi, X.; Hua, H.; Wu, Q.; Wang, X.; Wang, X.; Li, J. Effects of Different Feeding Methods on the Structure, Metabolism, and Gas Production of Infant and Toddler Intestinal Flora and Their Mechanisms. Nutrients 2022, 14. [Google Scholar] [CrossRef]
- Zang, Y.; Lai, X.; Li, C.; Ding, D.; Wang, Y.; Zhu, Y. The Role of Gut Microbiota in Various Neurological and Psychiatric Disorders-An Evidence Mapping Based on Quantified Evidence. Mediators of inflammation 2023, 2023, 5127157. [Google Scholar] [CrossRef]
- Xing, D.; Ren, N.; Li, Q.; Lin, M.; Wang, A.; Zhao, L. Ethanoligenens harbinense gen. nov., sp. nov., isolated from molasses wastewater. International journal of systematic and evolutionary microbiology 2006, 56, 755–760. [Google Scholar] [CrossRef]
- Konikoff, T.; Gophna, U. Oscillospira: a Central, Enigmatic Component of the Human Gut Microbiota. Trends in microbiology 2016, 24, 523–524. [Google Scholar] [CrossRef]
- Chen, Y.R.; Zheng, H.M.; Zhang, G.X.; Chen, F.L.; Chen, L.D.; Yang, Z.C. High Oscillospira abundance indicates constipation and low BMI in the Guangdong Gut Microbiome Project. Scientific reports 2020, 10, 9364. [Google Scholar] [CrossRef]
- Yang, J.; Li, Y.; Wen, Z.; Liu, W.; Meng, L.; Huang, H. Oscillospira - a candidate for the next-generation probiotics. Gut microbes 2021, 13, 1987783. [Google Scholar] [CrossRef]
- Eeckhaut, V.; Machiels, K.; Perrier, C.; Romero, C.; Maes, S.; Flahou, B.; Steppe, M.; Haesebrouck, F.; Sas, B.; Ducatelle, R. , et al. Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut 2013, 62, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. The New England journal of medicine 2020, 383, 2652–2664. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Kabir, I.; Tietelman, G.; Huan, C.; Fan, J.; Worgall, T.; Jiang, X.C. Sphingolipid de novo biosynthesis is essential for intestine cell survival and barrier function. Cell death & disease 2018, 9, 173. [Google Scholar] [CrossRef]
- Rajilić-Stojanović, M.; de Vos, W.M. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiology Reviews 2014, 38, 996–1047. [Google Scholar] [CrossRef]
- Nagao-Kitamoto, H.; Kamada, N. Host-microbial Cross-talk in Inflammatory Bowel Disease. Immune network 2017, 17, 1–12. [Google Scholar] [CrossRef]
- Hill-Burns, E.M.; Debelius, J.W.; Morton, J.T.; Wissemann, W.T.; Lewis, M.R.; Wallen, Z.D.; Peddada, S.D.; Factor, S.A.; Molho, E.; Zabetian, C.P. , et al. Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Movement disorders : official journal of the Movement Disorder Society 2017, 32, 739–749. [Google Scholar] [CrossRef]
- Rowin, J.; Xia, Y.; Jung, B.; Sun, J. Gut inflammation and dysbiosis in human motor neuron disease. Physiological reports 2017, 5. [Google Scholar] [CrossRef]
- Brenner, D.; Hiergeist, A.; Adis, C.; Mayer, B.; Gessner, A.; Ludolph, A.C.; Weishaupt, J.H. The fecal microbiome of ALS patients. Neurobiology of aging 2018, 61, 132–137. [Google Scholar] [CrossRef]
- Henke, M.T.; Kenny, D.J.; Cassilly, C.D.; Vlamakis, H.; Xavier, R.J.; Clardy, J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc Natl Acad Sci U S A 2019, 116, 12672–12677. [Google Scholar] [CrossRef]
- Ponzo, V.; Fedele, D.; Goitre, I.; Leone, F.; Lezo, A.; Monzeglio, C.; Finocchiaro, C.; Ghigo, E.; Bo, S. Diet-Gut Microbiota Interactions and Gestational Diabetes Mellitus (GDM). Nutrients 2019, 11. [Google Scholar] [CrossRef]
- Lee, G.; You, H.J.; Bajaj, J.S.; Joo, S.K.; Yu, J.; Park, S.; Kang, H.; Park, J.H.; Kim, J.H.; Lee, D.H. , et al. Distinct signatures of gut microbiome and metabolites associated with significant fibrosis in non-obese NAFLD. Nat Commun 2020, 11, 4982. [Google Scholar] [CrossRef] [PubMed]
- Munukka, E.; Rintala, A.; Toivonen, R.; Nylund, M.; Yang, B.; Takanen, A.; Hänninen, A.; Vuopio, J.; Huovinen, P.; Jalkanen, S. , et al. Faecalibacterium prausnitzii treatment improves hepatic health and reduces adipose tissue inflammation in high-fat fed mice. The ISME journal 2017, 11, 1667–1679. [Google Scholar] [CrossRef] [PubMed]
- Ai, D.; Pan, H.; Li, X.; Gao, Y.; Liu, G.; Xia, L.C. Identifying Gut Microbiota Associated With Colorectal Cancer Using a Zero-Inflated Lognormal Model. Frontiers in microbiology 2019, 10, 826. [Google Scholar] [CrossRef] [PubMed]
- Meehan, C.J.; Beiko, R.G. A phylogenomic view of ecological specialization in the Lachnospiraceae, a family of digestive tract-associated bacteria. Genome biology and evolution 2014, 6, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yu, D.; Wu, D.; Gao, X.; Shao, F.; Zhao, M.; Wang, J.; Ma, J.; Wang, W.; Qin, X. , et al. Tissue-resident Lachnospiraceae family bacteria protect against colorectal carcinogenesis by promoting tumor immune surveillance. Cell host & microbe 2023, 31, 418–432. [Google Scholar] [CrossRef]
- Ćesić, D.; Lugović Mihić, L.; Ozretić, P.; Lojkić, I.; Buljan, M.; Šitum, M.; Zovak, M.; Vidović, D.; Mijić, A.; Galić, N. , et al. Association of Gut Lachnospiraceae and Chronic Spontaneous Urticaria. Life (Basel, Switzerland) 2023, 13. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.; Cao, J.; Dong, Y.; Chen, Y. Gut microbiota-derived metabolites mediate the neuroprotective effect of melatonin in cognitive impairment induced by sleep deprivation. Microbiome 2023, 11, 17. [Google Scholar] [CrossRef]
- Takeuchi, T.; Kameyama, K.; Miyauchi, E.; Nakanishi, Y.; Kanaya, T.; Fujii, T.; Kato, T.; Sasaki, T.; Tachibana, N.; Negishi, H. , et al. Fatty acid overproduction by gut commensal microbiota exacerbates obesity. Cell metabolism 2023, 35, 361–375. [Google Scholar] [CrossRef]
- Duar, R.M.; Lin, X.B.; Zheng, J.; Martino, M.E.; Grenier, T.; Pérez-Muñoz, M.E.; Leulier, F.; Gänzle, M.; Walter, J. Lifestyles in transition: evolution and natural history of the genus Lactobacillus. FEMS Microbiol Rev 2017, 41, S27–s48. [Google Scholar] [CrossRef]
- Martín, R.; Miquel, S.; Ulmer, J.; Kechaou, N.; Langella, P.; Bermúdez-Humarán, L.G. Role of commensal and probiotic bacteria in human health: a focus on inflammatory bowel disease. Microbial cell factories 2013, 12, 71. [Google Scholar] [CrossRef]
- Yun, S.W.; Kim, J.K.; Lee, K.E.; Oh, Y.J.; Choi, H.J.; Han, M.J.; Kim, D.H. A Probiotic Lactobacillus gasseri Alleviates Escherichia coli-Induced Cognitive Impairment and Depression in Mice by Regulating IL-1β Expression and Gut Microbiota. Nutrients 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Asgharian, H.; Homayouni-Rad, A.; Mirghafourvand, M.; Mohammad-Alizadeh-Charandabi, S. Effect of probiotic yoghurt on plasma glucose in overweight and obese pregnant women: a randomized controlled clinical trial. European journal of nutrition 2020, 59, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.L.; Yu, H.Q.; Wu, R.J.; He, C.; Li, M.; Yan, H.; Li, J.J.; Wang, S.; Liu, Z.G.; Liu, Z.J. , et al. Thrombospondin 1 Modulates Monocyte Properties to Suppress Intestinal Mucosal Inflammation. Journal of innate immunity 2015, 7, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; He, G.Q.; Jia, J.L.; Zhu, Q.L.; Ruan, H. Oral administration of Clostridium butyricum for modulating gastrointestinal microflora in mice. Current microbiology 2011, 62, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; Peano, C.; Pass, D.A.; Foroni, E.; Severgnini, M.; Claesson, M.J.; Kerr, C.; Hourihane, J.; Murray, D.; Fuligni, F. , et al. Diversity of bifidobacteria within the infant gut microbiota. PLoS One 2012, 7, e36957. [Google Scholar] [CrossRef]
- Pinzone, M.R.; Celesia, B.M.; Di Rosa, M.; Cacopardo, B.; Nunnari, G. Microbial translocation in chronic liver diseases. International journal of microbiology 2012, 2012, 694629. [Google Scholar] [CrossRef]
Figure 1.
The forest map illustrates the results of Mendelian randomization (MR) analysis, indicating the impact of various gut microbiota genera on different lipid levels. A-F, the MR analysis demonstrates diverse effects of gut microbiota genera on HDL-C (A), LDL-C (B), TC (C), TG (D), APOA1 (E), APOB (F). nSNP, number of single nucleotide polymorphism; SE, standard error; OR, odds ratio; high-density lipoprotein cholesterol, HDL-C; low-density lipoprotein cholesterol, LDL-C; triglyceride, TG; total cholesterol, TC; apolipoprotein A1, APOA1; apolipoprotein B, APOB.
Figure 1.
The forest map illustrates the results of Mendelian randomization (MR) analysis, indicating the impact of various gut microbiota genera on different lipid levels. A-F, the MR analysis demonstrates diverse effects of gut microbiota genera on HDL-C (A), LDL-C (B), TC (C), TG (D), APOA1 (E), APOB (F). nSNP, number of single nucleotide polymorphism; SE, standard error; OR, odds ratio; high-density lipoprotein cholesterol, HDL-C; low-density lipoprotein cholesterol, LDL-C; triglyceride, TG; total cholesterol, TC; apolipoprotein A1, APOA1; apolipoprotein B, APOB.

Figure 2.
Scatter plot demonstrates a significant linear association between distinct gut microbiota genera and various forms of lipid metabolism within the human body, while no discernible heterogeneity of single nucleotide polymorphisms (SNPs) was observed. high-density lipoprotein cholesterol, HDL-C; low-density lipoprotein cholesterol, LDL-C; triglyceride, TG; total cholesterol, TC; apolipoprotein A1, APOA1; apolipoprotein B, APOB; SNP, single nucleotide polymorphisms..
Figure 2.
Scatter plot demonstrates a significant linear association between distinct gut microbiota genera and various forms of lipid metabolism within the human body, while no discernible heterogeneity of single nucleotide polymorphisms (SNPs) was observed. high-density lipoprotein cholesterol, HDL-C; low-density lipoprotein cholesterol, LDL-C; triglyceride, TG; total cholesterol, TC; apolipoprotein A1, APOA1; apolipoprotein B, APOB; SNP, single nucleotide polymorphisms..

Figure 3.
A comprehensive overview of the distribution of gut microbiota genera at the phylum and family levels, highlighting the subsequent influence on lipid levels. The red triangle represents the increasing effect, while the blue triangle represents the reducing effect. high-density lipoprotein cholesterol, HDL-C; low-density lipoprotein cholesterol, LDL-C; triglyceride, TG; total cholesterol, TC; apolipoprotein A1, APOA1; apolipoprotein B, APOB.
Figure 3.
A comprehensive overview of the distribution of gut microbiota genera at the phylum and family levels, highlighting the subsequent influence on lipid levels. The red triangle represents the increasing effect, while the blue triangle represents the reducing effect. high-density lipoprotein cholesterol, HDL-C; low-density lipoprotein cholesterol, LDL-C; triglyceride, TG; total cholesterol, TC; apolipoprotein A1, APOA1; apolipoprotein B, APOB.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.


A Two-Sample Mendelian Randomization Analysis Investigates Associations Between Gut Microbiota and Dyslipidemia
Xuyi Zhou
et al.
,
2023



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated