
Preprint
Article
iPSC-Derived Endothelial Cells Reveals LDLR-Dysfunction and Dysregulated Gene Expression Profiles in Familial Hypercholesterolemia
Altmetrics
Downloads
130
Views
64
Comments
0
A peer-reviewed article of this preprint also exists.
supplementary.zip (31.22KB )




Submitted:
08 November 2023
Posted:
08 November 2023
You are already at the latest version
Alerts
Abstract
Defects in low-density lipoprotein receptor (LDLR) are associated with familial hypercholesterolemia (FH), manifested by atherosclerosis and cardiovascular disease. Defective LDLR in hepatocytes leads to increased blood cholesterol level that damage vascular cells, especially endothelial cells, through oxidative stress and inflammation. However, the distinctions between endothelial cells from individuals with normal and defective LDLR are not yet fully comprehended. In this study, we obtained and examined endothelial derivatives of induced pluripotent stem cells (iPSC) generated previously from conditionally healthy donors and compound heterozygous FH patients carrying pathogenic LDLR alleles. In normal iPSC-derived endothelial cells (iPSC-EC), we detected the LDLR protein predominantly in its mature form, while iPSC-EC from patients with FH display reduced level of mature LDLR and show abolished low-density lipoprotein uptake. RNA-seq of iPSC-EC with mutant LDLR revealed a unique transcriptome profile comprising downregulated genes related to monocarboxylic acid transport, exocytosis and cell adhesion, as well as upregulated signaling pathways of cell secretion and leukocyte activation. Overall, these findings suggest that LDLR defects increases susceptibility of endothelial cells to inflammation and oxidative stress. This, combined with elevated extrinsic cholesterol levels, may result in accelerated endothelial dysfunction, contributing to early progression of atherosclerosis and other cardiovascular pathologies related to FH.
Keywords:
Subject: Biology and Life Sciences - Cell and Developmental Biology
1. Introduction
The low-density lipoprotein (LDL) receptor has a significant function in human lipid metabolism [1]. Numerous mutations within the LDLR gene are pathogenic, causing hereditary familial hypercholesterolemia (FH). Patients with FH have elevated blood cholesterol levels and concomitant lipid metabolism diseases, such as atherosclerosis, coronary heart disease, and Alzheimer’s disease.
The main role in the FH pathogenesis is assigned to the LDL receptors in liver cells, which are responsible for cholesterol uptake and metabolization. However, when the LDL receptors are defective or absent, hepatocytes have impaired low-density lipoprotein absorption which ultimately leads to increased cholesterol levels in the bloodstream. Elevated blood cholesterol indirectly affects blood vessels and vascular cells through the lipid accumulation inside the intima, causing oxidative stress, inflammatory cell recruitment and local cytokine production [2,3,4]. Nonetheless, it is still little known about how vascular endothelial cells themselves differ between individuals with normal and pathogenic LDLR alleles.
The LDL receptors are involved in the functioning of variety of cell types, including pluripotent progenitors, vascular smooth muscle cells, renal mesangial cells, renal tubular cells, podocytes, brain endothelial cells and umbilical artery [5]. It is believed that сellular metabolism and metabolites are crucial in regulating three-dimensional chromatin architecture and gene expression [6]. Therefore, we hypothesize that endothelial cells with dysfunctional LDLR and associated metabolic abnormalities in cellular metabolism might themselves have dysregulated gene expression profiles independent of excess extracellular cholesterol levels. Our study focused on identifying both the LDLR protein expression and transcriptomic patterns in endothelial cells with pathogenic allelic variants in the LDLR gene and discussed how this may potentially contribute to the FH progression. Previously, we obtained two lines of induced pluripotent stem cells (iPSCs) from patients who were compound heterozygotes for pathogenic and likely pathogenic allelic variants of the LDLR gene [7,8,9]. Both lines contained alterations within the LDLR gene that lead to impaired protein maturation, reducing the rate of LDLR transport to the membrane, and causing its partial retention in the endoplasmic reticulum. We obtained endothelial derivatives by direct differentiation of iPSC lines from both a conditionally healthy donor and patients with FH. We showed that typical endothelial derivatives of human iPSCs predominantly produce mature LDLR protein, whereas its level is significantly reduced in endothelial derivatives carrying pathogenic LDLR alleles. Transcriptomic analysis of FH patient-specific iPSC-derived endothelial cells (iPSC-ECs) revealed bias in gene expression in several signaling pathways. Cellular metabolic abnormalities caused by defective LDLR are manifested at the transcriptomic level as down-regulated expression of monocarboxylic acid transporters. Altered expression was also found for genes involved in exocytosis, cell-cell adhesion, cell secretion and leukocyte activation. These data suggest that endothelial cells with damaged LDLR are themselves more predisposed to dysfunction, oxidative stress, and chronic inflammation, which may be responsible for the earlier and accelerated progression of atherosclerosis and cardiovascular disease in patients with FH.
2. Results
2.1. Obtaining and characterization of endothelial derivatives from iPSCs
To study the properties of endothelial cells with pathogenic LDLR allelic variants, we obtained endothelial derivatives through directed differentiation from two iPSC lines FH 1.3.1S and FH 3.2.8T from patients with FH, as well as from iPSC lines K6-4f and K7-4Lf from a conditionally healthy donors. On day 8, we assessed the directed differentiation efficiency by quantifying VE-cadherin-positive cells by FACS. We found no significant differences between cultures of iPSC-ECs in the number of VE-cadherin-positive cells, which accounted for more than 90% of the differentiated derivatives (Figure 1A). This indicates that the endothelial differentiation of iPSCs from patients with FH was as efficient as the differentiation of iPSCs from a healthy donor. All resulting derivatives demonstrate an immunophenotype with the presence of the surface antigen CD31 and von Willebrand factor, attributable to endothelial cells (Figure 1B). In addition, iPSC-ECs with and without the LDLR mutation have similar ability to form capillary-like structures in Matrigel without significant differences in angiogenic potential parameters (Figure 1C). However, when we added fluorescently labelled low-density lipoprotein Dil-LDL to iPSC-ECs, we found that those from patients with FH were significantly reduced in ability to uptake the labelled LDL (Figure 2).
2.2. IPSCs and iPSC-derived endothelial cells (iPSC-EC) from patients with FH exhibit reduced levels of mature LDLR
LDL receptors have been found not only in hepatocytes, but also in the other cell types, including iPSCs and endothelium [20,21,22,23,24]. The results that showed us the ability of iPSC-ECs to uptake low-density lipoproteins indicate that obtained cells have LDL receptors. We further investigated the presence of mature and immature LDLR forms in protein extracts from iPSCs and iPSC-ECs with normal and pathogenic LDLR alleles using immunoblotting. As a control, we used HepG2 hepatocyte-like cells with properties of mature hepatocytes characteristics obtained from a patient with hepatocarcinoma [22]. We found that in iPSCs and iPSC-ECs with the native LDLR gene, predominantly mature LDLR is detected, as well as in hepatocyte-like HepG2 cells (Figure 3A-C). The relative LDLR protein levels in hepatocyte-like HepG2 cells, both normal iPSCs and iPSC-ECs did not differ significantly from each other. However, a significantly reduced level of the mature LDLR was found in iPSCs and iPSC-ECs from FH patients compared to normal iPSCs, their endothelial derivatives and hepatocyte-like HepG2 cell line (Figure 3A-C). The level of immature LDLR was significantly increased compared to the mature one in iPSC line FH 1.3.1 and its endothelial derivatives. In iPSC line FH 3.2.8 and its endothelial derivatives, both LDLR forms are detected at low levels (Figure 3A-C) and the total LDLR level is significantly reduced (Fig. 3C). In iPSC line FH 1.3.1 and its endothelial derivatives, the total LDLR level does not differ from that of conditionally healthy iPSCs and their endothelial derivatives (Figure 3B). Thus, we have shown that both iPSCs obtained from FH patients and their endothelial derivatives have reduced levels of mature LDLR.
2.3. Transcriptome profiling reveals dysregulation of various signaling pathways in LDLR mutant iPSC-ECs
We performed bulk RNA sequencing (RNA-seq) on two control iPSC-EC cultures (CTRL) and two LDLR mutant iPSC-EC cultures (FH). At least three technical replicates were used for each experiment. The PCA plot of the transcriptome of 12 samples showed a clustering of three technical replicates together and a distinct distribution in iPSC-EC cultures according to disease by dimension 2 (Figure 4A).
Direct comparison of total RNA expression of iPSC-ECs between CTRL and FH groups revealed 39 differentially expressed genes (DEGs), of which 26 were downregulated and 13 were upregulated in FH (logFC |1|, FDR<0.05; Figure 4 B, C, and Table S1). The top 5 down-regulated genes were MEG3, MEG8, FGB, MIR381HG, and HSALNG0039225 while the top 5 over-expressed genes were LINC01291, LINC02968, HSALNG0024559, IGLON5, and ILDR2 (Figure 4 B). The average expression level (counts per million) of the LDLR gene was 343 and 326 in FH and CTRL iPSC-EC lines respectively.
Gene ontology analysis of DEGs revealed several dysregulated biological pathways in LDLR mutant iPSC-EC lines (Figure 4D, Tables S2 and S3). Monocarboxylic acid transport (GO:0015718) genes (PLA2G5, SLC16A6, ADORA2B, and ABCC4) were downregulated in LDLR mutant iPSC-EC lines. Downregulated genes are also involved in the biological processes (BPs) of regulating exocytosis (GO:0017157; ADORA2B, FGB, STXBP6, PLA2G5, ABCC4, and SEMA5A), cell-cell adhesion (GO:0098609; FGB, SPARCL1, STXBP6, and CNTN4), and positive regulation of the response to external stimuli (GO:0032103; ADORA2B, PLA2G5, SEMA5A, S100A14, and HLA-DQB1).
The 13 upregulated DEGs were participate in the BPs that control cell secretion (GO:0046903; XDH, SYT11, and ILDR2) and regulate leukocyte activation (GO:0002694; SYT11, SIRPA, and ILDR2).
Among these DEGs, three were related to angiogenesis (GO:0001936) genes such as XDH, SEMA5A, and SULF1. The expression of SEMA5A and SULF1 was downregulated in FH iPSC-ECs, whereas XDH was upregulated.
Reactome Gene Sets analysis demonstrated that downregulated DEGs such as HLA-DQB1, IFIT2, and UBA7 were related to the interferon signaling pathway (R-HSA-913531). In terms of KEGG analysis, the DEGs were mainly involved in the Rap1 signaling pathway (hsa04015), including downregulated ADORA2B, GNAO1, and SULF1 but upregulated EFNA2 gene.
STRING analysis revealed an interaction between LDLR and SYT11, CNTN4, SEMA5A at the protein level.
3. Discussion
The current trend in modeling of human diseases is to collect patient-specific iPSCs from individuals with hereditary pathologies and to elucidate the mechanisms of this pathology at the molecular and cellular levels using relevant cells obtained by iPSC differentiation [23]. Patient-specific iPSC-ECs are broadly used to model various disorders, including pulmonary arterial hypertension, Moyamoya disease, fibrodysplasia ossificans progressiva, Huntington’s disease, Kawasaki’s disease, type I diabetes mellitus, atrial or ventricular septal defects, pulmonary valve stenosis, cardiomyopathy, calcified aortic valve disease, hemophilia A, diabetic endotheliopathy, Hutchinson-Gilford Progeria Syndrome, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, and peripheral artery disease [27,28,29,30,31,32].
There are known lines of iPSCs from patients with FH and examples of their use to model of this pathology, which have only been carried out on hepatocyte-like derivatives of iPSCs [20,21,22,33,34]. Despite the knowledge that endothelial cells are the main target in atherosclerosis and the progression of cardiovascular pathology in FH, ECs or iPSC-derived ECs from patients with FH have not yet been obtained or studied. Current studies of endothelial dysfunction in FH patients have been based on blood biomarkers, blood flow and microvascular function indicators [35,36,37].
In endothelial cells, LDLR is thought to be involved in the endocytosis and transcytosis of circulating LDLs [38,39,40]. LDLR is found expressed in the blood-brain barrier endothelium [35] and in human umbilical artery endothelial cells (HUAECs) [21]. However, we found no information on the LDLR expression in endothelial cells from FH patients, including iPSC-ECs. With this work, we fill this gap and hope that studying patient-specific iPSC-ECs may potentially contribute to the understanding of the FH pathogenesis and aid in the development of new treatment approaches.
In our study, we were the first to obtain iPSC-ECs from compound heterozygous patients with FH and demonstrated dysfunction of the LDLR protein. We have shown that both iPSCs and iPSC-ECs with pathogenic LDLR alleles, have significantly reduced levels of the mature form of LDLR and an increased level of the immature form compared to controls with normal LDLR.
The mutation in one LDLR allele of the FH 1.3.1 patient is c. 530С>T, leading to the substitution p.Ser177Leu (ClinVar ID 3686), which is considered pathogenic and classified as a transport-defective mutation [41,42]. The mutation in the other LDLR allele of the patient is c.1054T>C, resulting in p.Cys352Arg (ClinVar ID 251618), which is considered likely pathogenic and associated with a disruption in the EGF-like domain of the LDLR protein [38]. The second mutation can also be classified as transport-defective mutation [39].
In patient FH 3.2.8, an extended deletion c.2141-966_2390-330del spans introns 14-16 and exons 15-16 and, according to bioinformatic predictions, is considered pathogenic or disease causing as it leads to the loss of the O-glycosylation domain, which interferes with the production of the mature LDLR protein. The second mutation c.1327T>C leads to p.Trp443Arg (ID 998052), referred as likely pathogenic, causes a recycling defect when ligands are not released from the complex with LDLR in endosomes and, as a result, LDLR does not recirculates to the cell surface. Our results on LDLR expression allow us to conclude that all LDLR mutations in the iPSCs and iPSC-EC lines lead to the reduction in the mature form of the LDLR protein, while the combination of mutations in the second patient significantly reduces both mature and immature LDLR forms.
Pharmacological blocking of endoplasmic reticulum-associated degradation (ERAD) or using pharmacological chaperones are able to provide transport of immature LDLR to the cell membrane surface, restoring receptor function despite its incomplete maturation [36]. Thus, iPSC-derived ECs obtained from patients with FH may potentially help in the development and screening of targeted drugs to treat the consequences of specific mutations in patients with FH.
In this study, we also compared iPSC-ECs with normal and defective LDLR at the transcriptome level. We found no difference in LDLR gene expression between FH and CTRL iPSC-ECs, suggesting that LDLR function is mainly abolished at the post-translational level, as also shown by immunoblotting results.
Transcriptome profiling revealed the downregulation of the most genes (67%) in LDLR mutant iPSC-EC. We found that biological processes related to monocarboxylic acid transport, exocytosis and cell-cell adhesion were downregulated in LDLR mutant iPSC-EC lines. On the contrary, cell secretion and leukocyte activation were upregulated in these cells. Thus, the quantitative reduction of mature LDLR in iPSC-ECs abolished endothelial function, which was manifested both in the decreased ability to uptake LDL and at the transcriptomic level.
The down-regulated expression of monocarboxylic acid transporters likely reflects the expected dysregulation of cellular metabolism in iPSC-ECs with defective LDLR at the transcriptome level. Quantitative variations in metabolites associated with these transporters, including Ac-KoA and fatty acids (organic anions), may affect chromatin and gene regulation, while decreased levels of antioxidant transport could potentially render cells more susceptible to oxidative stress [45,46,47].
There was also dysregulation of angiogenesis genes (XDH, SEMA5A, and SULF1) and the Rap1 signalling pathway (ADORA2B, GNAO1, SULF1, EFNA2), which promotes endothelial homeostasis and may be involved in endothelial dysfunction-associated cardiovascular pathologies [43]. However, the iPSC-ECs with normal and defective LDLR showed no differences in the ability to form capillary-like structures in an angiogenesis test, both in terms of total vessel length and in the number of branching points. Therefore, it is likely that the identified dysregulation in angiogenesis genes may be related to other aspects of this process. It is also noteworthy that Rap1 signalling and the XDH gene are both related to oxidative stress [49,50].
Additionally, the reconstructed protein-protein interaction network showed an association between LDLR and some molecules such as SYT11, CNTN4, and SEMA5A. Interestingly, semaphorin and ephrin family members are involved in axon guidance and synaptic plasticity and they are also important in endothelial cell-leukocyte interactions during atherogenesis [46]. Synaptotagmin-11 (SYT11) and contactin 4 (CNTN4) are both essential for neural development [52,53]. SYT11 is a vesicle-trafficking protein that can suppress microglial activation by inhibiting cytokine secretion and phagocytosis [49]. CNTN4, a crucial adhesion protein, is involved in T-cell activation and oxLDL-induced cell apoptosis and inflammation in THP-1 macrophages [55,56]. Thus, our data suggest that dysregulation of neuronal guidance molecules (chemoattraction and chemorepulsion) can be considered as a possible mechanism for endothelial dysfunction in LDLR mutant iPSC-EC lines.
Overall, the transcriptomic analysis of iPSC-ECs with defective LDLR identified a number of genes and signaling pathways that could potentially be important for the progression of endothelial dysfunction in FH patients. Endothelial cells with damaged LDLR per se appear to be predisposed to dysfunction, oxidative stress and chronic inflammation, which may facilitate early and accelerated FH progression. However, further detailed studies are needed to confirm and understand our findings.
4. Materials and Methods
4.1. Ethical statements
4.2. iPSC lines and their cultivation
The study used two patient-specific iPSCs cell lines called FH 1.3.1S and FH 3.2.8T, hPSCregs numbers ICGi036-A and ICGi038-A [7,8]. They were obtained previously from FH patients being compound heterozygotes with pathogenic and likely pathogenic variants of both LDLR alleles, which were p.Ser177Leu / p.Cys352Arg for FH 1.3.1S and p.Glu714_Ile796del / p.Trp443Arg for FH 3.2.8T [7,8,9]. The control was the iPSC lines K7-4Lf and K6-4f from healthy donors, hPSCreg number ICGi022-A; ICGi021-A respectively [10]. The iPSCs were cultured in E8 medium (Thermo Fisher Scientific, Waltham, MA, USA) on a surface coated with Matrigel matrix (Corning, New York, USA) in an incubator at 37 °C and 5% CO2. During re-plating, cells were disaggregated with 0.5 mM EDTA and seeded at a 1:4 ratio into E8 medium supplemented with 10 mM Rho kinase inhibitor thiazovivine (Merck, Darmstadt, Germany).
4.3. Directed differentiation of iPSCs into endothelial derivatives
Directed differentiation of iPSCs obtained from a healthy donor and patients with FH in the endothelial direction was carried out according to the protocol [11] with our modifications [12]. The protocol includes three stages: mesodermal differentiation, endothelial differentiation and selection of endothelial derivatives by magnetic sorting. At the first stage (mesodermal differentiation), iPSCs were seeded in the E8 medium on a surface treated with Matrigel (Corning) to achieve 60-70% cell confluence the next day. On the second day, we washed the iPSCs from the E8 medium with PBS and added new medium containing RPMI 1640 (Thermo Fisher Scientific), 6 μM Chir99021 (Selleckchem, Houston, TX, USA), 1% B27 supplement without insulin and 100 units/mL penicillin-streptomycin to initiate mesodermal differentiation. On the 3rd day, the culture medium was completely replaced with a similar one, including RPMI 1640 (Thermo Fisher Scientific), 1% B27 without insulin, 100 units/mL penicillin-streptomycin, with the concentration of Chir99021 halved to 3 μM. The endothelial differentiation stage was performed from day 5 to day 8. The growth medium from the previous step was completely removed, the cells were washed with PBS and EGM-2 complete endothelial medium (Lonza) supplemented with 50 ng/mL VEGF, 25 ng/mL bFGF (all from Sci-store, Moscow, Russia), 10 μM SB431542 (R&D Systems, NE Minneapolis, MN, USA) was added. At the final stage of endothelial differentiation (day 9), the culture was enriched by magnetic sorting, selecting cells based on the CD31 marker of mature endothelial cells. Magnetic beads conjugated to anti-CD31 antibodies (Miltenyi Biotec, Bergisch Gladbach, Germany) were used for sorting according to the manufacturer’s protocol. Prior to incubation with magnetic beads, cells were disaggregated using Accutase enzyme (Thermo Fisher Scientific) and passed through a 0.70 μm cell strainer to remove conglomerates. Sorted CD31-positive cells were seeded in complete endothelial medium EGM-2 on a culture surface coated with type 4 collagen (Merck). Endothelial derivatives were passaged every 6–7 days using TrypLE Express (Thermo Fisher Scientific) at a ratio of 1:2 – 1:3 depending on cell density.
4.4. Flow cytometry
Quantitative detection of the endothelial marker VE-cadherin (CD144) in cultures after differentiation was carried out by flow cytometry using a FACS Aria III instrument at the Center for Analysis of Biological Objects in the Institute of Cytology and Genetics, SB RAS. We used anti-VE-cadherin (CD144) antibody (BD) and an isotype control mouse IgG1 (BD, Franklin Lakes, New Jersey , USA), both conjugated to the PerCP-Cy5.5 fluorochrome (Table 1). Cells were disaggregated using Accutase (Thermo Fisher Scientific) to avoid damage to the cell membrane containing the surface marker. Staining of endothelial derivatives with antibodies and appropriate isotype controls was performed according to the manufacturer’s protocol (BD). For one reaction, 105 cells were used in 100 μL of 1% BSA in PBS with the addition of 5 μL of antibodies or 2 μL of isotype control. The experiment was performed in three biological and three technical replicates. In each experiment, 104 events were counted.
4.5. Immunostaining
The immunostaining procedure was described in detail previously [13,14,15]. Briefly, differentiated endothelial derivatives were fixed with 4% PFA (Merck) for 10 min, permeabilized with 0.5% Triton X-100 (Merck) for 30 min and then blocked with 1% bovine serum albumin (BSA) in PBS. Incubation with primary antibodies was carried out overnight at +4 0C. Secondary antibodies were incubated with cells for 1.5 hours in the dark at room temperature. Cell nuclei were stained with DAPI. Imaging was performed on an inverted fluorescence microscope Ti-E (Nikon, Tokyo, Japan) using NIS Elements Advanced Research software. A list of primary and secondary antibodies is given in Table 1.
4.6. Functional examination of endothelial cells
To evaluate angiogenic potential, we polymerized 200 μL of Matrigel (Corning) diluted 1:1 with EGM-2 medium (Lonza, Basel, Switzerland) in a 1 cm2 well for 15 minutes at 37 °C. A suspension of 2x 105 endothelial cells with EGM-2 medium was then evenly distributed over the well surface and placed the cells in an incubator at 37 °C. Capillary-like structures formed by endothelial cells were detected after 4 hours on a Ti-E inverted fluorescence microscope (Nikon) using NIS Elements Advanced Research software. The parameters of capillary-like structures formed by endothelial cells were assessed using the AngioTool program [16].
LDL uptake capacity was assessed by adding fluorescently labeled low-density lipoprotein Dil-LDL (Thermo Fisher Scientific) to proliferating endothelial cells. Cells were incubated in serum-free medium for 24 hours prior to staining. Staining was performed overnight in serum-free medium supplemented with Dil-LDL at a concentration of 5 μg/mL. Imaging was performed using a Ti-E inverted fluorescence microscope (Nikon) and NIS Advanced Research software. To quantify fluorescence intensity, we captured cells in 10 fields of view at the same exposure value of 400 ms (Texas Red channel) and processed the images in the ImageJ program (https://ij.imjoy.io/).
4.7. Immunoblotting
Proteins were extracted from 105 cells in RIPA buffer (Merk). Extract of HepG2 cells were received from the Collective Center of ICG SB RAS “Collection of Pluripotent Human and Mammalian Cell Cultures for Biological and Biomedical Research” (https://ckp.icgen.ru/cells/; http://www.biores.cytogen.ru/brc_cells/collections/ICG_SB_RAS_CELL). Each sample of 8 μg was separated by 10% SDS-PAGE using a Tetra Mini-Protein Electrophoresis BIORAD system (Bio-Rad Lab, Berkeley, CA, USA). Recombinant human LDLR (R&D) was used as a control. The separated proteins were transferred from the gel to a PVDF membrane (Bio-Rad) using a Mini Trans-Blot wet transfer system (Bio-Rad Lab). The membrane was divided into two parts, guided by a pre-stained protein molecular weight marker (Bio-Rad Lab). Each part of the membrane was precipitated with antibodies; one against the target protein LDLR and the other against the reference protein ACTB. Secondary antibodies against goat and rabbit IgG conjugated to horseradish peroxidase were used for detection. The primary and secondary antibodies and their dilutions are shown in Table 1. Chemiluminescent signal detection was performed using the Bio-Rad Clarity Max Western ECL Substrate kit on a Bio-Rad ChemiDoc MP instrument. Densitometric analysis to measure differences in protein levels was performed using ImageJ software (https://ij.imjoy.io/). Based on the measurement results, the ratio of signal intensities between the target and reference proteins was obtained. We carried out each immunoblotting experiment in triplicate. Box plots construction and statistical comparisons were performed using the Wilcoxon method with Bonferroni correction for multiple comparisons in the R package (R version 4.2.0; R Core Team (2022). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/).
4.8. RNA-seq
For RNA sequencing, iPSC-EC cultures were lysed with Trizol reagent and then total RNA was extracted with a PureLink RNA micro kit (Invitrogen, Carlsbad, CA, USA). RNA quality was checked using BioAnalyser and RNA 6000 Nano Kit (Agilent, Santa Clara, CA, USA). Samples with RNA integrity number >8.0 were used for cDNA library preparation. PolyA RNA was purified from total RNA samples with Dynabeads® mRNA Purification Kit (Thermo Fisher Scientific). Illumina cDNA library was made from polyA NEBNext® Ultra™ II RNA Library Prep (New England BioLabs, Ipswich,MA, USA ) according to the manual. The resulting cDNA library was sequenced on the Illumina HiSeq 1500 platform at 75 bp read length by Genoanalytica (https://www.genoanalytica.ru). At least 30 million of read pairs were generated for each sample. Raw RNA-seq data that support the findings of this study are available from the corresponding author, upon reasonable request.
4.9. Statistics
Statistical data processing was carried out using the Wilcoxon test with Bonferroni correction for multiple comparisons. Statistical calculations and boxplots were obtained using the R package version 4.2.0 (R version 4.2.0; R Core Team (2022). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/).
RNA sequencing data were processed using DRAGEN Bio-IT v.3.9.5 (Illumina) and aligned to a reference human hg38 genome. The quality of the RNA-seq data was assessed using MultiQC v.1.11 [17]. Identification of differentially expressed genes (DEGs) between two control iPSC-EC cultures (CTRL) and two LDLR mutant iPSC-EC cultures (FH) was carried out using the EdgeR tool [18] with the following criteria: (i) log fold change ≥|1| and (ii) FDR adjusted p.value <0.05. Genes with negative fold change values were termed downregulated, while those with positive fold change values were termed upregulated. To analyze the gene ontology terms and pathways affected in the LDLR mutant iPSC-ECs, both downregulated and upregulated DEGs were used as input in the online server Metascape [19]. The protein-protein interaction network of LDLR and DEGs was constructed using the STRING database (https://string-db.org).
5. Conclusions
Overall, the transcriptomic analysis of iPSC-ECs with defective LDLR identified a number of genes and signaling pathways that could potentially be important for the progression of endothelial dysfunction in FH patients. Endothelial cells with damaged LDLR per se appear to be predisposed to dysfunction, oxidative stress and chronic inflammation, which may facilitate early and accelerated FH progression. However, further detailed studies are needed to confirm and understand our findings.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Differentially expressed genes between CTL and LDLR mutants; Table S2: Metascape enrichment downregulated genes in LDLR mutants; Table S3: Metascape enrichment upregulated genes in LDLR mutants.
Author Contributions
Conception and design: ISZ, AIS, AAS, MSN. Preparation of experiments, analysis and interpretation of data: ISZ, AIS, AMA, AAS, MSN, AAZ, SBS, NAT, VAS, NVZ. Contribution of reagents/materials/analysis tools/administrative and financial support: YVP, VVK, MVE, SMZ. Manuscript writing: ISZ, AIS, AAS; MSN. All authors read and approved the final manuscript.
Funding
This research was funded by the Russian Science Foundation grant No. 21-15-00065.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Local Biomedical Ethics Committee at the National Medical Research Center of Cardiology Ministry of Health of the Russian Federation, Moscow (protocol #246, 29 April 2019).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Raw data are available from the corresponding author upon request.
Acknowledgments
The immunofluorescent imaging and FACS was performed using resources of the Common Facilities Center of Microscopic Analysis of Biological Objects, ICG SB RAS (https://ckp.icgen.ru/ckpmabo/), supported by the State project of the Institute of Cytology and Genetics # 0259-2021-0011.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Goldstein, J.L.; Brown, M.S. The LDL Receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Kobiyama, K.; Ley, K. Atherosclerosis a Chronic Inflammatory Disease with an Autoimmune Component. Circ. Res. 2018, 123, 1118–1120. [Google Scholar] [CrossRef] [PubMed]
- Botts, S.R.; Fish, J.E.; Howe, K.L. Dysfunctional Vascular Endothelium as a Driver of Atherosclerosis: Emerging Insights Into Pathogenesis and Treatment. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ma, K.L.; Ruan, X.Z.; Liu, B.C. Dysregulation of the Low-Density Lipoprotein Receptor Pathway Is Involved in Lipid Disorder-Mediated Organ Injury. Int. J. Biol. Sci. 2016, 12, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Han, X.; Powell, C.A. Lipids and Genes: Regulatory Roles of Lipids in RNA Expression. Clin. Transl. Med. 2022, 12, e863. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, I.S.; Shevchenko, A.I.; Tmoyan, N.A.; Elisaphenko, E.A.; Kalinin, A.P.; Sleptcov, A.A.; Nazarenko, M.S.; Ezhov, M. V.; Kukharchuk, V. V.; Parfyonova, Y. V.; et al. Induced Pluripotent Stem Cell Line ICGi037-A, Obtained by Reprogramming Peripheral Blood Mononuclear Cells from a Patient with Familial Hypercholesterolemia Due to Heterozygous p.Trp443Arg Mutations in LDLR. Stem Cell Res. 2022, 60. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, I.S.; Shevchenko, A.I.; Tmoyan, N.A.; Elisaphenko, E.A.; Zubkova, E.S.; Sleptcov, A.A.; Nazarenko, M.S.; Ezhov, M. V.; Kukharchuk, V. V.; Parfyonova, Y. V.; et al. Induced Pluripotent Stem Cell Line ICGi036-A Generated by Reprogramming Peripheral Blood Mononuclear Cells from a Patient with Familial Hypercholesterolemia Caused Due to Compound Heterozygous p.Ser177Leu/p.Cys352Arg Mutations in LDLR. Stem Cell Res. 2022, 59, 102653. [Google Scholar] [CrossRef] [PubMed]
- Nazarenko, M.S.; Sleptcov, A.A.; Zarubin, A.A.; Salakhov, R.R.; Shevchenko, A.I.; Tmoyan, N.A.; Elisaphenko, E.A.; Zubkova, E.S.; Zheltysheva, N. V.; Ezhov, M. V.; et al. Calling and Phasing of Single-Nucleotide and Structural Variants of the LDLR Gene Using Oxford Nanopore MinION. Int. J. Mol. Sci. 2023, 24, 4471. [Google Scholar] [CrossRef] [PubMed]
- Malakhova, A.A.; Grigor’eva, E. V.; Pavlova, S. V.; Malankhanova, T.B.; Valetdinova, K.R.; Vyatkin, Y. V.; Khabarova, E.A.; Rzaev, J.A.; Zakian, S.M.; Medvedev, S.P. Generation of Induced Pluripotent Stem Cell Lines ICGi021-A and ICGi022-A from Peripheral Blood Mononuclear Cells of Two Healthy Individuals from Siberian Population. Stem Cell Res. 2020, 48. [Google Scholar] [CrossRef]
- Gu, M. Efficient Differentiation of Human Pluripotent Stem Cells to Endothelial Cells. Curr. Protoc. Hum. Genet. 2018, 98, e64. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.I.; Arssan, A.M.; Zakian, S.M.; Zakharova, I.S. Chemokine CCL2 Activates Hypoxia Response Factors Regulating Pluripotency and Directed Endothelial Differentiation of Human Pluripotent Stem Cells. Russ. J. Dev. Biol. 2023, 54, 134–146. [Google Scholar] [CrossRef]
- Zakharova, I.S.; Zhiven’, M.K.; Saaya, S.B.; Shevchenko, A.I.; Smirnova, A.M.; Strunov, A.; Karpenko, A.A.; Pokushalov, E.A.; Ivanova, L.N.; Makarevich, P.I.; et al. Endothelial and Smooth Muscle Cells Derived from Human Cardiac Explants Demonstrate Angiogenic Potential and Suitable for Design of Cell-Containing Vascular Grafts. J. Transl. Med. 2017, 15, 54. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, I.; Saaya, S.; Shevchenko, A.; Stupnikova, A.; Zhiven’, M.; Laktionov, P.; Stepanova, A.; Romashchenko, A.; Yanshole, L.; Chernonosov, A.; et al. Mitomycin-Treated Endothelial and Smooth Muscle Cells Suitable for Safe Tissue Engineering Approaches. Front. Bioeng. Biotechnol. 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Vaskova, E.A.; Medvedev, S.P.; Sorokina, A.E.; Nemudryy, A.A.; Elisaphenko, E.A.; Zakharova, I.S.; Shevchenko, A.I.; Kizilova, E.A.; Zhelezova, A.I.; Evshin, I.S.; et al. Transcriptome Characteristics and X-Chromosome Inactivation Status in Cultured Rat Pluripotent Stem Cells. Stem Cells Dev. 2015, 24, 2912–2924. [Google Scholar] [CrossRef] [PubMed]
- Zudaire, E.; Gambardella, L.; Kurcz, C.; Vermeren, S. A Computational Tool for Quantitative Analysis of Vascular Networks. PLoS One 2011, 6, e27385. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lun, A.T.L.; Smyth, G.K. From Reads to Genes to Pathways: Differential Expression Analysis of RNA-Seq Experiments Using Rsubread and the EdgeR Quasi-Likelihood Pipeline. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Caron, J.; Pène, V.; Tolosa, L.; Villaret, M.; Luce, E.; Fourrier, A.; Heslan, J.M.; Saheb, S.; Bruckert, E.; Gómez-Lechón, M.J.; et al. Low-Density Lipoprotein Receptor-Deficient Hepatocytes Differentiated from Induced Pluripotent Stem Cells Allow Familial Hypercholesterolemia Modeling, CRISPR/Cas-Mediated Genetic Correction, and Productive Hepatitis C Virus Infection. Stem Cell Res. Ther. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Catar, R.; Chen, L.; Zhao, H.; Wu, D.; Kamhieh-Milz, J.; Lücht, C.; Zickler, D.; Krug, A.W.; Ziegler, C.G.; Morawietz, H.; et al. Native and Oxidized Low-Density Lipoproteins Increase the Expression of the LDL Receptor and the LOX-1 Receptor, Respectively, in Arterial Endothelial Cells. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Aden, D.P.; Fogel, A.; Plotkin, S.; Damjanov, I.; Knowles, B.B. Controlled Synthesis of HBsAg in a Differentiated Human Liver Carcinoma-Derived Cell Line. Nat. 1979 2825739 1979, 282, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, S.P.; Shevchenko, A.I.; Zakian, S.M. Induced Pluripotent Stem Cells: Problems and Advantages When Applying Them in Regenerative Medicine. Acta Naturae 2010, 2, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.C.; Brown, E.E.; Abutaleb, N.O.; Truskey, G.A. Development and Application of Endothelial Cells Derived From Pluripotent Stem Cells in Microphysiological Systems Models. Front. Cardiovasc. Med. 2021, 8, 625016. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Wang, X.; Rivera-Bolanos, N.; Ameer, G.A. Generation of Autologous Vascular Endothelial Cells for Patients with Peripheral Artery Disease. J. Cardiovasc. Transl. Res. 2023. [Google Scholar] [CrossRef] [PubMed]
- Omer, L.; Hudson, E.A.; Zheng, S.; Hoying, J.B.; Shan, Y.; Boyd, N.L. CRISPR Correction of a Homozygous Low-Density Lipoprotein Receptor Mutation in Familial Hypercholesterolemia Induced Pluripotent Stem Cells. Hepatol. Commun. 2017, 1, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Omer, L.; Hindi, L.; Militello, G.; Stivers, K.B.; Tien, K.C.; Boyd, N.L. Familial Hypercholesterolemia Class II Low-Density Lipoprotein Receptor Response to Statin Treatment. DMM Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef]
- Okada, H.; Nakanishi, C.; Yoshida, S.; Shimojima, M.; Yokawa, J.; Mori, M.; Tada, H.; Yoshimuta, T.; Hayashi, K.; Yamano, T.; et al. Function and Immunogenicity of Gene-Corrected IPSC-Derived Hepatocyte-Like Cells in Restoring Low Density Lipoprotein Uptake in Homozygous Familial Hypercholesterolemia. Sci. Rep. 2019, 9, 4695. [Google Scholar] [CrossRef]
- Ramakrishnan, V.M.; Yang, J.Y.; Tien, K.T.; McKinley, T.R.; Bocard, B.R.; Maijub, J.G.; Burchell, P.O.; Williams, S.K.; Morris, M.E.; Hoying, J.B.; et al. Restoration of Physiologically Responsive Low-Density Lipoprotein Receptor-Mediated Endocytosis in Genetically Deficient Induced Pluripotent Stem Cells. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, A.; Moreira, A.S.B.; Muccillo, F.B.; Assad, M.; Tibirica, E. V. Microvascular Function and Endothelial Progenitor Cells in Patients with Severe Hypercholesterolemia and the Familial Hypercholesterolemia Phenotype. Cardiol. 2017, 137, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Pajkowski, M.; Dudziak, M.; Chlebus, K.; Hellmann, M. Assessment of Microvascular Function and Pharmacological Regulation in Genetically Confirmed Familial Hypercholesterolemia. Microvasc. Res. 2021, 138, 104216. [Google Scholar] [CrossRef] [PubMed]
- Vuorio, A.; Kovanen, P.T.; Raal, F.J. Coronary Microcirculatory Dysfunction in Hypercholesterolemic Patients with COVID-19: Potential Benefit from Cholesterol-Lowering Treatment. Ann. Med. 2023, 55, 2199218. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, F.; Crinelli, R.; Nasoni, M.G.; Benedetti, S.; Palma, F.; Fraternale, A.; Iuliano, L. LDL Receptors, Caveolae and Cholesterol in Endothelial Dysfunction: OxLDLs Accomplices or Victims? Br. J. Pharmacol. 2021, 178, 3104–3114. [Google Scholar] [CrossRef] [PubMed]
- Catar, R.; Chen, L.; Zhao, H.; Wu, D.; Kamhieh-Milz, J.; Lücht, C.; Zickler, D.; Krug, A.W.; Ziegler, C.G.; Morawietz, H.; et al. Native and Oxidized Low-Density Lipoproteins Increase the Expression of the LDL Receptor and the LOX-1 Receptor, Respectively, in Arterial Endothelial Cells. Cells 2022, Vol. 11, Page 204 2022, 11, 204. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, B.; Fenart, L.; Dehouck, M.P.; Pierce, A.; Torpier, G.; Cecchelli, R. A New Function for the LDL Receptor: Transcytosis of LDL across the Blood-Brain Barrier. J. Cell Biol. 1997, 138, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Oommen, D.; Kizhakkedath, P.; Jawabri, A.A.; Varghese, D.S.; Ali, B.R. Proteostasis Regulation in the Endoplasmic Reticulum: An Emerging Theme in the Molecular Pathology and Therapeutic Management of Familial Hypercholesterolemia. Front. Genet. 2020, 11, 570355. [Google Scholar] [CrossRef] [PubMed]
- Thormaehlen, A.S.; Schuberth, C.; Won, H.H.; Blattmann, P.; Joggerst-Thomalla, B.; Theiss, S.; Asselta, R.; Duga, S.; Merlini, P.A.; Ardissino, D.; et al. Systematic Cell-Based Phenotyping of Missense Alleles Empowers Rare Variant Association Studies: A Case for LDLR and Myocardial Infarction. PLOS Genet. 2015, 11, e1004855. [Google Scholar] [CrossRef] [PubMed]
- Semenova, A.E.; Sergienko, I. V.; García-Giustiniani, D.; Monserrat, L.; Popova, A.B.; Nozadze, D.N.; Ezhov, M. V. Verification of Underlying Genetic Cause in a Cohort of Russian Patients with Familial Hypercholesterolemia Using Targeted Next Generation Sequencing. J. Cardiovasc. Dev. Dis. 2020, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Garcia, U.; Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Larrea-Sebal, A.; Alonso-Estrada, R.; Aguilo-Arce, J.; Ostolaza, H.; Palacios, L.; Martin, C. Mutation Type Classification and Pathogenicity Assignment of Sixteen Missense Variants Located in the EGF-Precursor Homology Domain of the LDLR. Sci. Rep. 2020, 10, 1727. [Google Scholar] [CrossRef] [PubMed]
- Dahan, P.; Lu, V.; Nguyen, R.M.T.; Kennedy, S.A.L.; Teitell, M.A. Metabolism in Pluripotency: Both Driver and Passenger? J. Biol. Chem. 2019, 294, 5420–5429. [Google Scholar] [CrossRef] [PubMed]
- González-Becerra, K.; Ramos-Lopez, O.; Barrón-Cabrera, E.; Riezu-Boj, J.I.; Milagro, F.I.; Martínez-López, E.; Martínez, J.A. Fatty Acids, Epigenetic Mechanisms and Chronic Diseases: A Systematic Review. Lipids Health Dis. 2019, 18. [Google Scholar] [CrossRef] [PubMed]
- Trefely, S.; Lovell, C.D.; Snyder, N.W.; Wellen, K.E. Compartmentalised Acyl-CoA Metabolism and Roles in Chromatin Regulation. Mol. Metab. 2020, 38. [Google Scholar] [CrossRef]
- Chrzanowska-Wodnicka, M. Rap1 in Endothelial Biology. Curr. Opin. Hematol. 2017, 24, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Remans, P.H.J.; Gringhuis, S.I.; van Laar, J.M.; Sanders, M.E.; Papendrecht-van der Voort, E.A.M.; Zwartkruis, F.J.T.; Levarht, E.W.N.; Rosas, M.; Coffer, P.J.; Breedveld, F.C.; et al. Rap1 Signaling Is Required for Suppression of Ras-Generated Reactive Oxygen Species and Protection Against Oxidative Stress in T Lymphocytes. J. Immunol. 2004, 173, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Baek, B.S.; Song, S.H.; Kim, M.S.; Huh, J.I.; Shim, K.H.; Kim, K.W.; Lee, K.H. Xanthine Dehydrogenase/Xanthine Oxldase and Oxidative Stress. J. Am. Aging Assoc. 1997, 20, 127–140. [Google Scholar] [CrossRef]
- Van Gils, J.M.; Ramkhelawon, B.; Fernandes, L.; Stewart, M.C.; Guo, L.; Seibert, T.; Menezes, G.B.; Cara, D.C.; Chow, C.; Kinane, T.B.; et al. Endothelial Expression of Guidance Cues in Vessel Wall Homeostasis Dysregulation under Proatherosclerotic Conditions. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Shimojo, M.; Madara, J.; Pankow, S.; Liu, X.; Yates, J.; Südhof, T.C.; Maximov, A. Synaptotagmin-11 Mediates a Vesicle Trafficking Pathway That Is Essential for Development and Synaptic Plasticity. Genes Dev. 2019, 33, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Oguro-Ando, A.; Bamford, R.A.; Sital, W.; Sprengers, J.J.; Zuko, A.; Matser, J.M.; Oppelaar, H.; Sarabdjitsingh, A.; Joëls, M.; Burbach, J.P.H.; et al. Cntn4, a Risk Gene for Neuropsychiatric Disorders, Modulates Hippocampal Synaptic Plasticity and Behavior. Transl. Psychiatry 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Wang, Y.; Zhang, F.; Yan, S.; Guan, Y.; Gong, X.; Zhang, T.; Cui, X.; Wang, X.; Zhang, C.X. Synaptotagmin-11 Inhibits Cytokine Secretion and Phagocytosis in Microglia. Glia 2017, 65, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Huang, X.-T.; Miao, Y.-P.; Bai, X.-L.; Jin, F. MiR-148a-3p Attenuates Apoptosis and Inflammation by Targeting CNTN4 in Atherosclerosis. Ann. Transl. Med. 2022, 10, 1201–1201. [Google Scholar] [CrossRef] [PubMed]
- Jeon, B.-N.; Kim, S.; Kim, Y.; Yu, H.; Kim, H.; Ha, Y.; Kim, Y.Y.; Park, C.; Kim, G.; Cha, M.; et al. CNTN4/APP Axis of Cancer Cells and T-Cells. Res. Sq. 2023, preprint. [Google Scholar] [CrossRef]
Figure 1.
Characteristics of endothelial derivatives obtained by differentiating iPSCs from patients with FH and a healthy donor. (a) VE-cadherin quantification of iPSC-derived EC from a healthy donor (K7-4) and patients with FH (FH 1. 3.1S and FH 3.2.8T). (b) IPSC-EC from a healthy donor and patients with FH are positively stained with antibodies to CD31 (red) and von Willebrand factor (green). Cell nuclei are stained with DAPI. The scale bar is 100 microns. (c) Capillary-like structures formed by endothelial derivatives of iPSCs from a healthy donor (EC K7-4) and patients with FH (FH EC 3.1S and FH EC 3.2.8T). Angiogenic potential quantification on total and average vasculature length, and total number of branching points.
Figure 1.
Characteristics of endothelial derivatives obtained by differentiating iPSCs from patients with FH and a healthy donor. (a) VE-cadherin quantification of iPSC-derived EC from a healthy donor (K7-4) and patients with FH (FH 1. 3.1S and FH 3.2.8T). (b) IPSC-EC from a healthy donor and patients with FH are positively stained with antibodies to CD31 (red) and von Willebrand factor (green). Cell nuclei are stained with DAPI. The scale bar is 100 microns. (c) Capillary-like structures formed by endothelial derivatives of iPSCs from a healthy donor (EC K7-4) and patients with FH (FH EC 3.1S and FH EC 3.2.8T). Angiogenic potential quantification on total and average vasculature length, and total number of branching points.

Figure 2.
iPSCs-derived endothelium from patients with FH show reduced LDL uptake capacity. Representative images showing differences in the fluorescently labeled LDL uptake capacity (red signal) between endothelial derivatives from iPSCs with normal (EC K7-4) and pathological (EC FH 1.3.1 and EC FH 3.2.8) LDLR allelic variants. Nuclei are stained with DAPI. The scale bar is 100 microns. Quantification of LDL uptake capacity for control and patient-specific IPSC-derived endothelial cells.
Figure 2.
iPSCs-derived endothelium from patients with FH show reduced LDL uptake capacity. Representative images showing differences in the fluorescently labeled LDL uptake capacity (red signal) between endothelial derivatives from iPSCs with normal (EC K7-4) and pathological (EC FH 1.3.1 and EC FH 3.2.8) LDLR allelic variants. Nuclei are stained with DAPI. The scale bar is 100 microns. Quantification of LDL uptake capacity for control and patient-specific IPSC-derived endothelial cells.
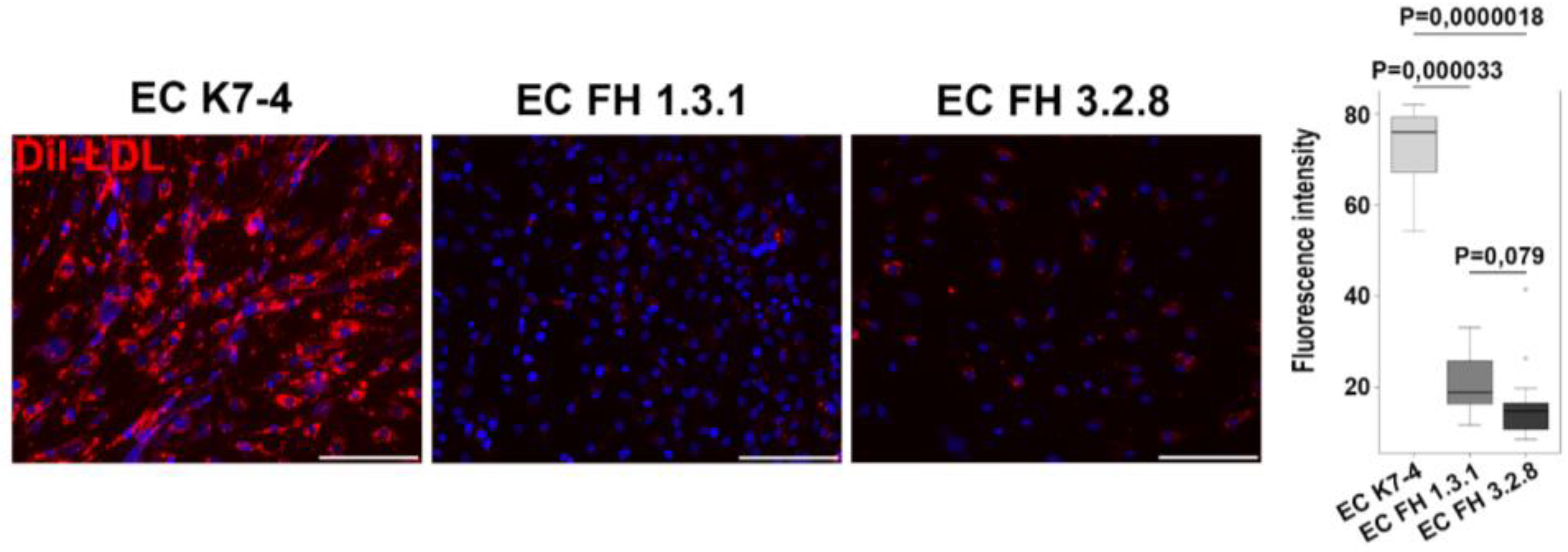
Figure 3.
LDLR protein expression in iPSCs and their endothelial derivatives from patients with FH and a healthy donor. (a) Immunoblotting revealed a decrease in the level of mature LDLR in iPSCs and iPSC-ECs from patients with FH (FH 1.3.1 and FH 3.2.8), as well as an increased level of immature LDLR in iPSCs and iPSC-ECs from FH 1.3.1. compared with this in a healthy person (K7-4). (b) Relative densitometric quantification of total LDL (mature and immature forms together) in iPSCs and iPSC-ES from patients with FH (FH 1.3.1 and FH 3.2.8) and a healthy donor (K7-4). (c) Relative densitometric quantification of mature and immature LDL in iPSCs and iPSC-ES from patients with FH (FH 1.3.1 and FH 3.2.8) and a healthy donor (K7-4).
Figure 3.
LDLR protein expression in iPSCs and their endothelial derivatives from patients with FH and a healthy donor. (a) Immunoblotting revealed a decrease in the level of mature LDLR in iPSCs and iPSC-ECs from patients with FH (FH 1.3.1 and FH 3.2.8), as well as an increased level of immature LDLR in iPSCs and iPSC-ECs from FH 1.3.1. compared with this in a healthy person (K7-4). (b) Relative densitometric quantification of total LDL (mature and immature forms together) in iPSCs and iPSC-ES from patients with FH (FH 1.3.1 and FH 3.2.8) and a healthy donor (K7-4). (c) Relative densitometric quantification of mature and immature LDL in iPSCs and iPSC-ES from patients with FH (FH 1.3.1 and FH 3.2.8) and a healthy donor (K7-4).

Figure 4.
Transcriptional profiling of the control (CTRL) and LDLR mutant (FH) iPSC-EC. (A) PCA plot for two control iPSC-EC lines (CTRL) and two LDLR mutant iPSC-EC lines (FH). (B) Volcano plot of 39 differentially expressed genes (DEGs) between CTRL and FH. FDR, false discovery rate; FC, multiple shifts. The top 5 DEGs are shown. (C) Heat map of 39 DEGs. The color of each dot represents the gene expression in the sample. The brighter the red, the higher the expression; the brighter the blue, the lower the expression. (D) Histograms showing gene ontology of biological processes and reactome gene sets with upregulated (logFC ≥ 1) and downregulated (logFC ≤ −1) expression in FH compared to CTRL. (E) Bioinformatics analysis of DEGs using STRING to identify functional interactions between deregulated proteins. Each node represents a protein and each edge represents an interaction.
Figure 4.
Transcriptional profiling of the control (CTRL) and LDLR mutant (FH) iPSC-EC. (A) PCA plot for two control iPSC-EC lines (CTRL) and two LDLR mutant iPSC-EC lines (FH). (B) Volcano plot of 39 differentially expressed genes (DEGs) between CTRL and FH. FDR, false discovery rate; FC, multiple shifts. The top 5 DEGs are shown. (C) Heat map of 39 DEGs. The color of each dot represents the gene expression in the sample. The brighter the red, the higher the expression; the brighter the blue, the lower the expression. (D) Histograms showing gene ontology of biological processes and reactome gene sets with upregulated (logFC ≥ 1) and downregulated (logFC ≤ −1) expression in FH compared to CTRL. (E) Bioinformatics analysis of DEGs using STRING to identify functional interactions between deregulated proteins. Each node represents a protein and each edge represents an interaction.


Table 1.
Primary and secondary antibodies.
| Name | Supplier | Catalogue number |
RRID | Isotype | Dilution |
|---|---|---|---|---|---|
| FACS | |||||
| CD144+ PerCP-Cy5.5 | BD | 561566 | AB_10715835 | mouse IgG1 | 1/20 |
| mouse IgG1 + PerCP-Cy5.5 | BD | 552834 | AB_394484 | mouse IgG1 | 1/50 |
| Immunofluorescence | |||||
| CD 31 | Cell Marque | 131M-96 | AB_1516765 | mouse IgG1 | 1/50 |
| Von Willebrand factor | Dako | A0082 | AB_2315602 | rabbit IgG | 1/200 |
| mouse IgG1 + Alexa 568 | Thermo Fisher Scientific |
A21124 | AB_2535766 | goat IgG | 1/400 |
| rabbit IgG + Alexa 488 | Thermo Fisher Scientific |
A110088 | AB_143165 | goat IgG | 1/400 |
| goat IgG + Alexa 488 | Thermo Fisher Scientific |
A11055 | AB_2534102 | donkey IgG | 1/400 |
| Immunoblotting | |||||
| LDLR | R&D System | AF2148 | AB_2135126 | goat IgG | 1/1000 |
| ACTB | Abcam | ab8227 | AB_2305186 | rabbit IgG | 1/5000 |
| goat IgG + peroxidase | Jackson ImmunoResearch |
705-035-003 | AB_2340390 | donkey IgG | 1/5000 |
| rabbit IgG + peroxidase | Jackson ImmunoResearch |
711-035-152 | AB_10015282 | donkey IgG | 1/5000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.


iPSC-Derived Endothelial Cells Reveals LDLR-Dysfunction and Dysregulated Gene Expression Profiles in Familial Hypercholesterolemia
Irina S. Zakharova
et al.
,
2023



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated