
Preprint
Article
HIF-1α/MMP-9 Axis Is Required for the Early Phases of Skeletal Myoblast Differentiation under Normoxia Condition In Vitro
Altmetrics
Downloads
94
Views
31
Comments
0
A peer-reviewed article of this preprint also exists.


This version is not peer-reviewed
Submitted:
07 November 2023
Posted:
08 November 2023
You are already at the latest version
Alerts
Abstract
Hypoxia-inducible factor (HIF)-1α represents the oxygen-sensitive subunit of HIF transcriptional factor, usually degraded in normoxia and stabilized in hypoxia to regulate several target gene expression. Nevertheless, in the skeletal muscle satellite stem cells (SCs), an oxygen level-independent regulation of HIF-1α has been observed. Although HIF-1α has been highlighted as SC function regulator, its spatio-temporal expression and role during myogenic progression remain controversial. We herein analyzed HIF-1α expression and localization in differentiating murine C2C12 myoblasts and SCs under normoxia and evaluated matrix metalloproteinase (MMP)-9 as a HIF-1α target, by biomolecular, biochemical, morphological and electrophysiological analyses. HIF-1α expression increased after 24/48 h of differentiating culture and tended to decline after 72 h/5 days. Committed and proliferating mononuclear myoblasts exhibited nuclear HIF-1α expression, differently from the more elongated and parallel-aligned cells, likely ready to fuse with each other, which show mainly a cytoplasmic localization of the factor. Multinucleated myotubes displayed both a nuclear and cytoplasmic HIF-1α expression. The MMP-9 and MyoD (myogenic activation marker) expression synchronized with that of HIF-1α, increasing after 24 h of differentiation. Short-interfering RNA silencing of HIF-1α and MMP-9 and MMP-9 pharmacological inhibition unraveled MMP-9 as HIF-1α downstream effector and HIF-1α/MMP-9 axis essential for the morpho-functional cell myogenic commitment.
Keywords:
Subject: Biology and Life Sciences - Cell and Developmental Biology
1. Introduction
Postnatal mammalian skeletal muscle growth, maintenance and regeneration after damage is mostly dependent on the activity of satellite cells (SCs), the resident stem cell population [1,2]. In healthy muscle, these mononucleated cells are located in a specialized niche at the periphery of adult striated multinucleated myofibers (hence the name) between the sarcolemma and the basal lamina ensheathing each myofiber, in a dormant state. In response to different biochemical and mechanical stimuli emanated by the surrounding growing or injured microenvironment, SCs become activated, exit the quiescent state and start to proliferate and differentiate essentially recapitulating embryonal myogenesis steps [3,4,5]. SCs are characterized by the expression of the paired box transcription factor Pax7, regarded as the canonical SC-specific marker required for their survival and functionality and by the expression of the myogenic regulatory factors (MRFs) Myf5, MyoD, MRF4 and myogenin. In particular, when quiescent, SCs express Pax7 and the myogenic transcription factor Myf5 (detectable in ~90% of quiescent SCs). Once activated, they undergo mitotic division to give rise to a progeny of proliferating adult myoblasts co-expressing Pax7, Myf5 and the myogenic determinant factor 1 (MyoD), the master regulator of SC proliferation/activation. Afterwards, most of these cells lose Pax7, maintain MyoD expression (Pax7+/MyoD+) and progress into the myogenic program acquiring myogenin and MRF4/Myf6 expression to differentiate into skeletal myocytes. These, in turn, fuse to one another forming myotubes and finally new myofibers. At the same time a small population maintains Pax7 and down-regulates MyoD (Pax7+/MyoD−) and self-renew, not executing the myogenic program and reconstituting a compartment of quiescent ‘stem cell’ able to respond to future demand [6]. It has been demonstrated that SCs, as other stem cells, undergo an energy metabolic reprogramming during differentiation, consisting of a shift in metabolic substrate utilization. In particular, it has been shown that proliferating myoblasts mainly depend on glycolytic energy production differently from myotubes that exhibit an increased aerobic capacity [7,8,9]. Recent studies have highlighted the Hypoxia-inducible factor (HIF)-1α as a regulator of myogenesis and SCs/myoblasts’ homeostasis in vitro and in vivo [10,11,12,13,14,15,16,17,18,19,20,21,22]. It is a dimeric basic helix-loop-helix-PER-ARNT-SIM transcriptional factor consisting of a constitutively expressed subunit HIF-1β and an oxygen-regulated/sensitive subunit HIF-1α (or its paralogs HIF-2α and HIF-3α). HIF-1α biological activity depends on post-transcriptional modifications that are responsible for the α-subunit stability and activity. Usually in normoxia, post translational modifications of HIF-1α (HIF-1α is hydroxylated by the active prolyl-hydroxylases -PHDs-) lead to the subunit degradation via ubiquitin-proteasome pathway. By contrast, in hypoxia PHDs are inhibited, HIF-1α cannot be hydroxylated and becomes stable. It translocates to the cell nucleus, where it forms a transcriptional complex with HIF-1β which is able to interact with different coactivators and to regulate the expression of several target genes involved in many cellular processes including regulation of glycolytic metabolism in skeletal muscle [15,23,24,25]. However, in skeletal muscle tissue and SCs an oxygen level-independent regulation of the expression and activation of HIF-1α has been observed thus suggesting a role of such a protein in skeletal muscle physiological functions and its homeostasis. Nevertheless some controversy remains on the spatio-temporal expression of HIF-1α and on its exact role in myoblasts during myogenic differentiation. In addition, the knowledge of molecular targets of this factor needs to be expanded [10,17,18,19,21,22,26,27,28,29,30,31,32,33].
The present study was undertaken to better clarify the role of HIF-1α in myoblasts undergoing myogenic differentiation under normoxia conditions and to evaluate MMP-9 as a downstream effector of HIF-1α. In particular, we investigated MMP-9 as target gene when considering that MMP-9 has been demonstrated to be a target of HIF-1α in different cell types [34,35,36,37] and the role of MMP-9 in skeletal myogenesis has been documented [38,39,40,41].
This research demonstrates that: i) the expression of HIF-1α synchronized with that of MMP-9 peaking at early phase of differentiation (24 h) concomitantly to that of MyoD ii) MMP-9 was a target of HIF1-α and iii) HIF-1α/MMP-9 axis was necessary for the morpho-functional myogenic activation/commitment of myoblasts.
2. Materials and Methods
2.1. Cell culture and treatments
Murine C2C12 skeletal myoblasts (American Type Culture Collection-ATCC; Manassas, VA, USA), were cultured in proliferation medium (PM) consisting in DMEM supplemented with 10% FBS (Sigma-Aldrich, Milan, Italy) and 1% penicillin/streptomycin (Sigma-Aldrich) at 37°C in a humidified incubator under 5% CO2 and 95% air. To induce myogenic progression the cells were cultured in PM until reaching 80% confluence and then shifted in myogenic differentiation medium (DM) consisting in DMEM supplemented with 2% horse serum (HS; Sigma-Aldrich) for different times (24, 48, 72 h and 5 days). In some experiments C2C12 cells were cultured in PM or in DM for 24 h in the presence of a MMP-9 inhibitor, SB-3CT (10 μM; Sigma-Aldrich), as previously reported [40].
Murine primary SCs were isolated from single myofibers of Extensor Digitorum Longus (EDL) of male Swiss adult mice (25-30 g) according to previously published protocol [5] (n = 3 animals; 24 myofibers for each muscle). Briefly, EDL muscles were digested in DMEM supplemented with 0.2% collagenase type I (Sigma-Aldrich). Single living myofibers were isolated from each others with a Pasteur pipette by means of a gentle mechanical trituration in serum-free culture medium and then transferred individually in a 24-well plate treated with Matrigel (BD Biosciences, San Jose, CA, USA). The myofibers were cultured for 48 h in the specific SC growth medium containing DMEM plus 20% FBS, 10% HS, 0.5% chicken embryo extract (Sera Laboratories International Ltd, Horsted Keynes, UK) and 1% penicillin/streptomycin, to allow the sprouting of SCs. Thereafter the myofibers were removed and the sprouted cells were cultured until 80% of confluence (3-5 days) before being detached with a solution of 0.05% trypsin–0.03% ethylenediaminetetraacetic acid (Sigma-Aldrich) and re-seeded in a culture plate for 20 min with fresh PM. After 20 min, the non-adherent cells (SCs) were collected and cultured on gelatin-coated glass coverslips in SC growth medium for 24 and 48 h and then processed for confocal immunofluorescence microscopy analysis.
2.2. Silencing of HIF-1a and MMP-9 by Short Interfering RNA
C2C12 cells seeded either on sterile glass coverslips put on the bottom of a well of a 6-well culture plate o directly on the well of a 6-well culture plate were cultured in PM till reaching 60-80% of confluence before being transfected with a pool of 3 short target-specific 20-25 nucleotides of interfering RNA duplexes (siRNA; 20 nM, Santa Cruz Biotechnology, Santa Cruz, CA, USA) designed to knock-down HIF-1α (sc-35562; Sense: 5’CACCAUGAUAUGUUUACUATT3’, Antisense: 5’UAGUAAACAUAUCAUGGUGTT3’, Sense: 5’CCAGUUGAAUCUUCAGAUATT3’, Antisense:5’UAUCUGAAGAUUCAACUGGTT3’; Sense: 5’CCACUUUGAAUCAAAGAAATT3’, Antisense: 5’UUUCUUUGAUUCAAAGUGGTT3’) or MMP-9 gene expression (sc-29401; Sense: 5’GCUUCCCUCUGAAUAAAGATT3’, Antisense: 5’UCUUUAUUCAGAGGGAAGCTT3’; Sense: 5’CUUCCAGUACCAAGACAAATT3’, Antisense: 5’UUUGUCUUGGUACUGGAAGTT3’; Sense: 5’CAGCUACUUUAGUCAAUCATT3’,Antisense: 5’UGAUUGACUAAAGUAGCUGTT3’). The cells transfected with a non-specific scrambled (SCR)-siRNA (20 nM, Santa Cruz Biotechnology, sc-37007) were used as control. siRNA transfections were carried essentially as previously published protocol [42], according to the manufacturer’s instructions. For reaching a higher efficiency of inhibition of HIF-1α expression two transfections with the specific siRNA were performed within 24 h of each other. After transfection - PM (T0) -, the cells were shifted in DM and cultured for 24 h before being processed for Western Blotting, immunofluorescence and electrophysiological analyses.
2.3. Morphological analyses
2.3.1. Phase Contrast microscopy
Myotube formation after 5 days of C2C12 cell culture in DM was evaluated under an inverted phase contrast microscopy (20X; Nikon Diaphot 300).
2.3.2. Confocal Laser scanning microscopy
Cells on glass coverslips were fixed with 0.5% PFA for 10 min at room temperature and processed for indirect immunostaining as previously reported protocol [43]. The employed primary antibodies (4°C; overnight) were indicated in Table 1.
The immunoreactions were revealed by incubation with specific secondary antibodies anti-rabbit or anti-mouse Alexa Fluor 488- or anti-mouse 568-conjugated IgG (1:200; Molecular Probes, Eugene, OR, USA) for 1 h at RT. In some experiments the cells were incubated with Alexa Fluor 488-labeled phalloidin (1:400 for 20 min at room temperature; Molecular Probes) to reveal F-actin filaments. Negative controls were carried out by replacing primary antibodies with non-immune serum; the cross-reactivity of the secondary antibodies was assessed by omitting the primary antibodies. The glass coverslips with the immunolabeled cells were mounted with an antifade mounting medium (Biomeda Gel mount; Electron Microscopy Sciences, Foster City, CA, USA). Observations were performed under a confocal Leica TCS SP5 microscope equipped with a HeNe/Ar laser source for fluorescence measurements and differential interference contrast (DIC) optics (Leica Microsystems, Mannheim, Germany) by using a Leica Plan Apo 63×/1.43NA oil immersion objective. Series of optical sections (1024 × 1024 pixels each; pixel size 204.3 nm) 0.4 μm in thickness were taken through the depth of the cells at intervals of 0.4 μm and projected onto a single ‘extended focus’ image. Quantitative analysis of Ki67- and MyoD- positive nuclei was performed by counting the positive cells in at least 5 random 200 × 200 μm2 square microscopic fields (63x objective) in each cell preparation and the results were reported as the percentage of positive nuclei on the total cell nuclei. Densitometric analyses of fluorescent signal intensity of each specific marker were performed on digitized images using ImageJ 1.49v software (http://rsbweb.nih.gov/ij) in 10 regions of interest (ROI) of 100 μm2 for each confocal stack (at least 10). The experiments were all performed in triplicate.
2.4. Total RNA Extraction and semi quantitative Reverse Transcription (RT)-PCR
Total RNA was extracted from C2C12 cells by using TRIzol Reagent (Invitrogen, Life Technologies, Grand Island, NY, USA), according to the manufacturer’s instructions. Reverse transcription and amplification of 1 µg of total RNA were performed in a thermal cycler (Perkin Elmer, Monza, Italy) using qScript XLT One-Step RT-PCR Kit (Quantabio, Beverly, Massachusetts, USA). After a starting cycle at 55 °C for 30 min and 94°C for 2 min for pre-denaturation, the samples were subjected to 40 cycles of PCR performed alternating at 94 °C for 15 s, 55 °C for 30 s and 72 °C for 1 min; the final extension step was carried on with a cycle at 72 °C for 5 min. The following mouse gene-specific primers were used: HIF-1α (NM_001313919.1), forward 5’-TCA GCA TAC AGT GGC ACT CA-3’ and reverse 5’-AAG GGA GCC ATC ATG TTC CA-3’ (transcript length 213 bp); MMP-9 (NM_013599.4), forward 5’-CTG GCG TGT GAG TTT CCA AA-3’ and reverse 5’-CTA GCA CCT TTC CCT CGG AT-3’ (transcript length 201 bp); β-actin (NM_007393.3), forward 5′-ACT GGG ACG ACA TGG AGA AG-3′ and reverse 5′- ACC AGA GGC ATA CAG GGA CA-3′ (transcript length 206 bp). β-actin mRNA was used as the internal control. Samples with no template (only water) were carried on in each run. The PCR products were separated on 1,8% agarose gel electrophoresis stained with ethidium bromide and detected with a UV transilluminator. Densitometric analysis of the bands was performed by using ImageJ 1.49v software (NIH, https://imagej.nih.gov/ij/) and normalized respect to β-actin.
2.5. Western Blotting
Total proteins were extracted from C2C12 cells grown in the different experimental conditions and quantified as previously reported [42]. Forty µg of total proteins were electrophoresed on NuPAGE® 4–12% Bis-Tris Gel (Invitrogen; 200 V, 40 min) and blotted onto polyvinylidene difluoride (PVDF) membranes (Invitrogen; 30 V, 1 h). The membranes were incubated on a rotary shaker first for 30 min at room temperature with the Blocking Solution included in the Western Breeze® Chromogenic Western Blot Immunodetection Kit (Invitrogen) and then overnight at 4°C with the antibodies reported in Table 1. Protein immunodetection was performed according to the Western Breeze®Chromogenic Immunodetection protocol (Invitrogen). Densitometric analysis of the bands was performed using ImageJ 1.49v software (https://imagej.nih.gov/ij/) and the values normalized to α-tubulin, assuming α-tubulin as control invariant protein, for each result.
2.6. Electrophysiological recordings
C2C12 cells cultured on glass coverslips - placed in standard plastic culture dishes (35 mm × 10 mm, Corning Incorporated, Corning, NY) - in PM or DM silenced or not for HIF-1α expression or in the presence or absence of SB-3CT (10 μM), were subjected to the electrophysiological analysis as previously reported [44,45]. Bioelectrical features of the cells were investigated by the whole cell patch clamp technique at room temperature (22°C).
The patch electrodes were produced from borosilicate glass capillaries (GC150-15; Clark, Electromedical Instruments, Reading, UK) by a two-step vertical puller (Narishige, Tokyo, Japan) and then filled with the following filling-pipette solution (mM): 130 KCl, 10 NaH2PO4, 0.2 CaCl2, 1 EGTA, 5 MgATP and 10 HEPES (pH 7.2 with KOH). When filled, electrode resistance was around 1.5–2 MΩ. The coverslip with the cells was positioned in the experimental chamber on the stage of an inverted microscope (Nikon Eclipse TE200, Amstelveen, NL) filled with the following physiological solution (mM): 150 NaCl, 5 KCl, 2.5 CaCl2, 1 MgCl2, 10 D-glucose and 10 HEPES (pH 7.4 with NaOH). The experimental apparatus for the electrophysiological records comprised an Axopatch 200 B amplifier, A/D-D/A interfaces Digidata 1200; Pclamp 6 software (Axon Instruments, Foster City, CA, USA) as described in earlier paper [46]. Data analysis was made by Clampfit 9 software (Axon Instruments, Foster City, CA, USA). The voltage-clamp mode of the Axopatch amplifier was set to analyze the membrane passive properties: two step voltage pulses of ± 10 mV were applied from a holding potential (HP) of −70 mV. We calculated the cell linear capacitance (Cm) of the patched cell from the area beneath the capacitive transient current. Since the membrane-specific capacitance is = 1 μF/cm2 this parameter can be considered as an index of the cell surface area. We also calculated the membrane conductance (Gm) from the steady-state membrane current (Im) as described in previous papers [46] to obtain the specific membrane conductance by the ratio Gm/Cm. Still in voltage-clamp mode we carried out the ion currents analysis. Ion currents were evoked by a suitable pulse protocol of stimulation consisting of 1-s step voltage pulses applied from a HP = −80 mV and ranging from −80 to 50 mV, in 10 mV increments. The capacitive and leak currents were eliminated on-line by the P4 procedure. For a correct comparison of the current values recorded from cells of different dimensions we normalized their amplitude to Cm and the resulting ratio I/Cm (expressed in pA/pF) represents the current density.
To better put in evidence the eventual occurrence of Ca2+ currents, without different overlapping ion fluxes, we filled the bath recording chamber with a Na+- and K+-free high-TEA external solution (mM): 10 CaCl2, 145 tetraethylammonium bromide, 10 HEPES and the patch pipette with a suitable internal solution (mM): 150 CsBr, 5 MgCl2, 10 Ethylene-bis(oxyethylenenitrilo) tetraacetic acid (EGTA), 10 (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) (pH = 7.2). All of the chemicals were acquired from Sigma-Aldrich.
2.7. Statistical analysis
Data are reported as mean ± SD of at least three independent experiments performed in triplicates. Statistical analysis was performed with one-way ANOVA with post-hoc Tukey HSD test calculator for comparing multiple treatments (https://astatsa.com/OneWay_Anova_with_TukeyHSD/) and p<0.05 was considered statistically significant. For the multiple comparisons of electrophysiological data we used One way ANOVA followed by Bonferroni's post hoc test.
3. Results
3.1. MMP-9 and HIF-1α expression during C2C12 myoblast differentiation under normoxia
The myogenic differentiation model of C2C12 cells was constructed and verified by both extensive morphological and biochemical analyses (Figure 1).
Accordingly, C2C12 cells were cultured in PM for 24 h and then shifted to DM for different times (24, 48, 72 h and 5 days) to observe the differentiation, always maintaining physiological oxygen levels. We observed that C2C12 cells after 24 h of culture in differentiation medium (DM, DMEM + 2% HS) exhibited nuclear expression of the proliferation marker Ki67 (Figure 1A,B,M) and of the myogenic commitment marker MyoD (Figure 1E,N). In particular the number of Ki67+ cells appeared reduced as compared to cells cultured in PM (Figure 1A). The cells cultured in PM did not express MyoD (data not shown). Moreover by using a specific antiserum recognizing both Notch-1 receptor and the Notch intracellular domain (Notch-ICD) which is proteolytically cleaved after Notch activation, a key determinant of satellite cell activation, proliferation and self-renewal [47,48] we observed that the cells showed an increased expression of Notch-1 (Figure 1I,J,O) in the cytoplasm and nucleus as compared with the cells in PM. The expression of such markers declined in the time (Figure 1A-G,I-O). After 72 h of culture the cells displayed nuclear expression of myogenin (Figure 1H) the myogenic regulatory factor in terminal differentiation. After 5 days they fuse with each other to form elongated multinucleated myotubes -with nuclei positive for myogenin- and expressing α-sarcomeric actin (Figure 1P-R), strongly indicative of cell myogenic differentiation.
Western Blotting analyses of SDH-B and LDH-A suggested that the cells underwent a metabolic shift during differentiation, from anaerobic glycolysis to oxidative pathway. Indeed we observed a decrease of the expression of LDH-A over time (Figure 2B) concomitant to an increase of SDH-B expression (Figure 2A).
In line with these observations, the cells exhibited also an increase of the expression of PGC-1α, recognised as a key regulator of mitochondrial biogenesis (Figure 2C-H), which reached the maximum expression level in the myotubes (Figure 2F,G).
Subsequently we investigated the expression and subcellular localization of HIF-1α during C2C12 myoblast differentiation by RT-PCR, Western Blotting and confocal immunofluorescence analyses (Figure 3).
HIF-1α mRNA expression remained constant in C2C12 cells both under growth and myogenic differentiating phases (Figure 3A). The protein expression was detected at each time point of differentiation with an increase after 24 h and 48 h of culture in myogenic DM and a tendency to decline after 72 h and 5 days (Figure 3B). Confocal immunofluorescence analysis revealed that HIF-1α was mainly expressed in the nucleus of proliferating and mononuclear myoblasts committed to the differentiation process (24 h). After 48 h when the cells were more elongated and parallel aligned, likely ready to fuse with each other, HIF-1α appeared mainly localized in the cytoplasm; in the latest phases of differentiation, myotubes displayed both a cytoplasmic and nuclear expression of HIF-1α (Figure 3C). In parallel, we analyzed the expression of MMP-9 at the mRNA and protein level and we found that it mostly synchronized with that of HIF-1α being higher in the early phase of myogenic differentiation and declining over the time (Figure 4A,B). Confocal immunofluorescence analysis revealed that MMP-9 resulted mainly localized in the cytoplasm along filamentous structures (Figure 4C-H).
To increase the definition of our findings, we analyzed the expression of HIF-1α and MMP-9 in primary murine SCs sprouted from single living myofibers isolated from EDL muscles (Figure 5).
Similarly, to what observed in C2C12 myoblasts (Figure 3C), Pax7+ SCs detected in the SC- enriched cultures, exhibited both nuclear and cytoplasmic expression of HIF-1α (Figure 5A) after 24 h of culture in their specific PM. After 48 h of culture (Figure 5B), multinucleated myotubes exhibited mainly a cytoplasmic expression of HIF-1α while showing, as expected, the downregulation of Pax7. Similarly to C2C12 myoblasts, SCs displayed a concomitant MMP-9 expression (Figure 5C,D).
3.2. HIF-1α and MMP-9 are required for myogenic commitment of myoblasts and MMP-9 is a downstream target of HIF-1α
3.2.1. Morphological and biochemical analyses
Based on the above finding concerning the temporal expression of HIF-1α and MMP-9 further analyses were focused on the early phase of cell differentiation (24 h). In particular, we evaluated the role of HIF-1α in the early phase of cell differentiation by silencing its expression by specific siRNA (PM T0) before culturing the cells in DM for 24 h (Figure 6A,C). As revealed by Western blotting analysis, silenced cells exhibited a reduced expression of MyoD (Figure 6A) and of Notch-ICD (Figure 6B) compared to not-silenced cells (wild type, WT and SCR-siRNA).
Confocal immunofluorescence analysis, while corroborating Western Blotting results related to MyoD and Notch-1 (Figure 7E-L), revealed also a reduction of the percentage of Ki67 positive nuclei when the cells were silenced for HIF-1α expression (Figure 7A-D). Notably as judged by DIC imaging, the silenced cells did not show those morphological features detectable in more advanced stages of differentiation such as alignment or fusion into myotubes (Figure 6D). These observations suggested that HIF-1α was required for cell commitment. Notably silenced cells exhibited a reduced expression of MMP-9, suggesting MMP-9 as downstream target of HIF-1α (Figure 6B, 7M-P).
Finally, to address the role of MMP-9 in the early phase of differentiation we performed experiments by inhibiting its expression by specific siRNA or its function by the pharmacological inhibitor SB-3CT (10 µM). We found that C2C12 cells silenced for MMP-9 (Figure 8A,F-I) after 24 h of culture in DM showed a reduction of Ki67 (Figure 8J-M,V), MyoD (Figure 8A,N-Q,W), Notch-1 (Figure 8A,R-U,X) and exhibited morphological features of not differentiated cells similarly to the cells silenced for HIF-1α (Figure 8B-D). Comparable results were obtained when the cells were cultured in DM in the presence of the MMP-9 inhibitor SB-3CT (Figure 8E,M,Q,U,X).
3.2.2. Functional electrophysiological analyses
To corroborate the results on the role of HIF-1α and MMP-9 on myogenic activation and commitment, we investigated some typical electrophysiological functional features of sarcolemma that better characterize an activated/committed myoblast (Figure 9).
Accordingly, we performed analyses on C2C12 cells cultured in PM or in DM for 24 h. In parallel with the morphological evaluation, the cells were transfected in the proliferation medium (PM) with either scrambled siRNA (SCR-siRNA) or HIF-1α-siRNA and then induced to differentiate by shifting in the differentiation medium (DM) for 24 h. Again, the not transfected C2C12 cells are indicated as WT. In other experiments the cells were cultured in PM or DM in the presence of the MMP-9 inhibitor SB-3CT (10 µM) for 24 h. In particular, we first considered the cell membrane capacitance (Cm) values (Figure 9B), obtained by the analysis of the current records evoked by the step pulse protocol shown in Figure 9A (bottom and top, respectively). While these values did not significantly change in any condition for cells grown in PM, we observed that Cm values recorded from WT C2C12 cells cultured in DM for 24 h, were significantly higher than those measured from WT cells in PM. This can suggest that after 24 h of differentiation we could already observe significant variations ascribable to cell surface enlargement. SCR-siRNA cells showed Cm values similar to WT. In contrast, the silencing of HIF-1α expression determined a significant reduction of Cm values compared to those recorded in WT or SCR-siRNA cells in DM, resulting not significantly different to those recorded in cell cultured in PM. The same effect was observed by blocking MMP-9 with the specific SB-3CT inhibitor. The specific membrane conductance Gm/Cm values (Figure 9C) that, in line with previous literature [49], tended to increase during the early phases of C2C12 cell differentiation, did not show statistically significant differences when cells were grown in PM in any condition tested. In contrast, WT C2C12 cells cultured in DM for 24 h showed a significant increase of the Gm/Cm compared to those in PM. Such values became again similar to PM when HIF-1α was silenced or MMP-9 was inhibited. These experiments, achieved on cells in the early phase of differentiation (DM 24 h), strongly indicate the involvement of HIF-1α and MMP-9 in determining the increase of Cm and Gm/Cm, typically observed under the differentiation. A summary of the different passive membrane properties is reported in Table 2.
Finally, by applying a suitable voltage pulse protocol of stimulation (Figure 10I), we evaluated the possible transmembrane ion current flux elicited by voltage-operated Ca2+ channels activation (ICa), as a typical feature of skeletal muscle differentiation.
WT C2C12 myoblasts grown in PM showed an inward almost not-inactivating current (Figure 10A) without a clear voltage threshold of appearance. Notably, when cells were kept in PM, we observed analogous inward current time courses for all of the conditions analyzed (Figure 10A-D).
The I-V plots related to all the experiments achieved on the cells grown in PM (Figure 10J) exhibited a current amplitude increase that was approximately linear with the applied voltage step increment, suggesting a scarcely voltage-dependent ion entry. Moreover, the mean current values were almost overlapping despite the different treatments. In contrast, WT cells cultured in DM for 24 h (Figure 10E) typically showed an inward current with a larger amplitude and a sort of inactivation at higher voltage steps, resembling the ICa time course of a more differentiated skeletal muscle cell. As expected, a similar time course was observed in SCR-siRNA (Figure 10F). Nonetheless, when HIF-1α was silenced, we observed current records (Figure 10G) with a time course quite different from those observed in WT cells in DM, but very similar to those recorded from cells in PM (Figure 10A) that varied linearly with the voltage step applied, as shown by the I-V plot in Figure 10K (triangles). This phenomenon was pretty similar to that evoked in WT myoblasts in PM, suggesting that, although in DM, HIF-1α-silenced cells still maintain the same ion channel profile/features shown by WT cells in PM. Finally, when MMP-9 was inhibited by SB-3CT (Figure 10H) the current time course recorded from cells in DM was quite different from the WT counterpart in the same medium, again resembling that observed in C2C12 WT in PM. Also the I-V plot supported this outcome (Figure 10K, diamonds) since data related to HIF-1α silencing and MMP-9 inhibitor were almost superimposed. In contrast, the mean values of the current amplitudes recorded from WT and SCR-siRNA tended to be higher and more similar. This result may further indicates that HIF-1α and MMP-9 have a role in driving ion channels expression during the early differentiation phase of C2C12 myoblasts.
4. Discussion
The current study provides experimental evidence that HIF-1α plays a critical function during myoblast differentiation in vitro that is not strictly related to hypoxic conditions. In fact, we have uncovered a previously unrecognized relationship between HIF-1α and MMP-9 in myoblasts induced to differentiate under normoxia conditions. In particular, we have evinced the essential role of such an axis in the very early phase of the normoxic myogenic progression, namely activation and commitment. These conclusions arise from the following observations.
First, morphological and biochemical analyses showed a different spatial and temporal expression of HIF-1α protein in the C2C12 cells undergoing differentiation: in particular HIF-1α expression increased after 24 h of culture in differentiating condition (DM) as compared to those observed in the cells in growing condition (PM) and declined with the differentiating time. Interestingly, HIF-1α expression is synchronized with that of myogenic activation marker MyoD and with Notch-1, a key determinant of myoblast activation and proliferation. Proliferating committed mononuclear myoblasts exhibited a more marked nuclear expression of HIF-1α as compared to the more elongated cells and polynucleated myotubes. The expression pattern of HIF-1α was confirmed on murine primary SCs. Conversely Wagatsuma et al. 2011 [30] found that C2C12 cells showed a decrease of HIF-1α protein expression when switching from growth to differentiation medium, whereas Ono et al. 2006 [21] showed that the HIF-1α protein was hardly detected in undifferentiated C2C12 myoblasts cultured in growth medium. This discrepancy may be attributed to different cell culture experimental conditions. However, according to our findings and other previous research [19,29], these papers [21,30] demonstrated the nuclear expression of HIF-1α in the early stage (induction) of C2C12 cell myogenesis process. In myoblasts the nuclear HIF-1α expression may be consistent with the functional induction of the genes encoding glycolytic enzymes required to satisfy the cell energy demand in the early phase of myogenic differentiation process [7,8,9,19,50,51,52]. Along this line, we have confirmed in our experimental myogenesis model that the cells undergo metabolic reprogramming during the differentiation process: in fact, proliferating myoblasts mainly depended on glycolytic energy production whereas myotubes showed a more developed aerobic capacity, as judged by the analyses of the expression of LDH-A and SDH-B. Moreover, during differentiation time we have observed an increase of the expression of PGC-1α that has been demonstrated to regulate LDH by decreasing LDH-A mRNA transcription and the enzymatic activity of pyruvate to lactate conversion [53]. In addition, PGC-1α has been recognized as a crucial regulator of mitochondrial biogenesis. Indeed, a clear consensus is evident in the literature on the occurrence of mitochondrial biogenesis during myogenic differentiation so as to sustain the energy demand required for myotube formation and successively polynucleated long myofibers [54]. Moreover, taking in mind the role of vascular endothelial growth factor (VEGF)/VEGF receptor-mediated signaling in myogenic differentiation [47,55,56,57] and the well-known cross-talk between HIF-1α and such signaling [19,58,59,60], we may also postulate the possibility that HIF-1α is physiologically involved in myoblast activation and proliferation by modulating VEGF signaling. Different from the protein expression, we have found that HIF-1α mRNA was constantly expressed in C2C12 myoblasts both under growth and differentiating steps according to previous data [19,21,29,30]. Our results indicate that the differences in protein level are not due to a different mRNA expression and suggest instead that the regulation of HIF-1α expression and activity during myogenic differentiation in vitro occurred at post-transcriptional level, possibly including protein transduction and stabilization/degradation, nuclear localization and transactivation.
The hypoxia-independent expression of HIF-1α in myoblastic cells during differentiation is not surprising as demonstrated in previous research [21,22,30]. This observation is also in line with the increasing evidence showing that HIF-1α is involved in different biological functions requiring its activation under normoxic conditions. The exact reason why HIF-1α escapes proteasome degradation in our cell model is not explained. However, non-hypoxic activators of HIF-1α have been described including, among others, nitric oxide NO [61,62] demonstrated to be produced by C2C12 myoblasts, peaking within 24 h after the induction of differentiation [63]. Moreover, the stability of HIF-1α in skeletal muscle cells depends on different regulators including not only the oxygen sensors named prolyl hydroxylases PHDs that guide the oxygen-dependent degradation of HIF-1α, but also the heat shock protein 90 (Hsp90) whose content and expression is independent on oxygen concentration. In particular, it has been observed that Hsp90 protects HIF-1α from proteasome degradation promoting its accumulation during in vitro differentiation of myoblastic cells under normoxia [21,22]. A PCG-1α dependent stabilization of HIF-1α has been also proposed in skeletal muscle cells [64].
Furthermore, the observed cytoplasmic expression of HIF-1α, may suggest an unconventional function of HIF-1α, as a signaling regulator distinct from its canonical activity as a transcriptional factor [65,66,67]. The specific mechanisms modulating HIF-1α expression needs further exploration.
The potential critical role of HIF-1α during the early phase of myogenesis under normoxia, suggested by our first results, has been then confirmed by experiments in which HIF-1α gene expression was silenced by specific siRNA. Indeed, silenced cells cultured in myogenic differentiation medium showed a reduction of the activation markers tested, namely Ki67, Notch-1 and MyoD. Taking in mind that during the process of myoblast differentiation, MyoD expression increases at first to decrease then [68], as confirmed in our experimental model, our results might also suggest a more differentiated state of the silenced cells and, in turn, the possibility that HIF-1α could function counteracting differentiation. However, morphological analysis uncovered that silenced cells did not show morphological features of more differentiated cells such as elongation, alignment or fusion into myotubes, leading us to conclude that HIF-1α is just required for cell activation/commitment. Our findings are consistent with previous data demonstrating that siRNA-mediated knockdown of HIF-1α in C2C12 myoblasts results in inhibiting myotube formation [21] and with data from Cirilli et al [15] showing that the pharmacological activation of HIF-1α by the treatment with some PHDs inhibitors promoted C2C12 myoblast differentiation. In contrast, it has been reported that the constitutive expression of active HIF-1α in C2C12 myoblasts did not accelerate differentiation such as the ectopic expression of DEC1/Stra13, a target gene of HIF-1α, did not prevent C2C12 myotube formation under either hypoxia or normoxia [69], supporting the notion that HIF-1α is not directly involved in postnatal myogenesis. Nevertheless, all these observations may suggest the requirement of a coordinated temporal modulation of HIF-1α and thus it cannot be excluded that: i) HIF-1α functionality may be critical just for some steps of the myogenic process, ii) HIF-1 may act differently during the differentiation period. Therefore, the above cited papers are not in contrast with the findings of the present study. However the role of HIF-1α in the late phase of myogenesis in our model remains to be investigated.
A novel finding from this study is the demonstration that MMP-9 is a downstream target of HIF-1α and that HIF-1α/MMP-9 signaling pathway is involved in myoblast activation. Indeed we found that MMP-9 expression synchronized with that of HIF-1α showing higher level after 24 h of differentiation, and that myoblasts silenced for HIF-1α exhibited a reduction of MMP-9 expression. Of note, the gene silencing of MMP-9 or its pharmacological inhibition caused a decrease of the activation markers of myogenesis similarly to HIF-1α silencing, thus suggesting an unconventional intracellular action of such proteases. In this line, a previous study conducted by our research group unraveled that bone marrow mesenchymal stromal cell secretome was able to increase MMP-9 expression in murine myoblasts and that this event was accompanied by an increase of cell mobilization, proliferation and activation [40]. Consistent with our findings, an earlier study demonstrated that inhibition of MMP-9 (with doxycycline and anti-MMP-9) delayed in vitro proliferation and differentiation of satellite cell- derived myoblasts from rat Soleus muscle [70]. The same research group documented that rat proliferating myoblasts exhibited a nuclear expression of MMP-9 and that MMP-9 activity was required for proper cell cycle progression, since its inhibition affected myoblast proliferation [38,41]. Different from Zimowska et al. we did not observe a nuclear localization of the protein suggesting that in our experimental model MMP-9 is not directly involved in nuclear events. Based on the observed distribution of MMP-9 that appeared linearly organized along filamentous structures, most likely cytoskeletal ones, we may hypothesize the occurrence of a MMP-9-dependent cytoskeletal rearrangement required for driving myoblast activation and differentiation [49,71,72].
A strength point of this research is that the outcomes from biomolecular, biochemical and morphological analyses, were robustly supported by the functional electrophysiological recordings. In particular, the analysis of the passive properties of the committed myoblasts focused on those parameters supposed to be indicative of a more differentiated phenotype. Accordingly, the cell capacitance (Cm), which is an index of the cell surface, is expected to increase during differentiation [73,74]. This was actually observed in our cells induced to differentiate, but when HIF-1α expression was silenced or MMP-9 was inhibited, its values turned out to be similar (not statistically different) to those measured in proliferating (not differentiating) myoblasts. Analogous observations could be made for the analysis of the specific conductance (Gm/Cm), that can be considered another index of differentiation [75]. Also preliminary results on resting membrane potential, that is supposed to become more polarized in myotubes than in myoblasts, confirmed these indications (data not shown). Finally, we intended to estimate the ion current flowing through voltage-activated Ca2+ channels. This, in fact, can be considered as a typical feature of skeletal muscle differentiation for its crucial role in excitation-contraction (EC) coupling in adult skeletal muscle fibers [76]. Although the structure of skeletal muscle L-type calcium channels consists of the pore forming α1 subunit, and the α2δ, β, and γ subunits, interestingly, these subunits are usually not expressed simultaneously during differentiation of myotubes in vitro [77,78,79]. This fact may undoubtedly hamper the early acquisition of fully functional voltage-dependent properties and their consequent clear revelation by electrophysiological techniques. Indeed, in our study we could observe that untreated myoblasts (WT) grown in PM usually showed an inward not-inactivating current suggesting a scarcely voltage-dependent ion entry. In contrast, although 24 h might be a very early differentiation stage, myoblasts cultured in DM exhibited an inward current with an increased amplitude and a sort of inactivation at higher voltage steps, resembling the ICa time course of a more differentiated skeletal muscle cell. Again, current recorded from cells silenced for HIF-1α was very similar to that evoked in WT myoblasts in PM, suggesting that, although in DM, HIF-1α-silenced cells maintain the same ion channel profile/features shown by WT in PM. A similar result was observed when MMP-9 was inhibited by SB-3CT. This may suggest that HIF-1α and MMP-9 have a role in C2C12 myoblasts driving Ca2+/ion channels expression/assembly during the early differentiation phase.
This hypothetical role of HIF-1α may be supported by previously published data even if achieved in a different muscle tissue, namely in pulmonary arterial smooth muscle cells [80]. The Authors reported that HIF-1α plays a key role in Ca2+ homeostasis, especially/specifically/just modulating the expression of transient receptor potential channels (TRPCs), thus contributing to pulmonary vascular remodeling.
5. Conclusions
In conclusion, our results highlight that HIF-1α plays a key role in normoxic differentiating skeletal myoblasts in vitro and corroborate the notion that MMP-9 is a downstream effector of this factor in myoblasts. We here demonstrate, for the first time to our knowledge, that the HIF-1α/MMP-9 axis is required for myoblast activation and commitment steps, thus broadening the understanding of the myogenic machinery. Deeper insight into the molecular pathways that drive and coordinate skeletal myogenesis is essential to improve the comprehension of skeletal muscle disorders and pathologies. Moreover, these results may provide cues for the identification of novel smart targets that could be exploited to boost the intrinsic regenerative potential of muscle tissue thus contributing to increase the therapeutic proposals in muscle disease management. Indeed, SCs are notoriously scarce in the muscle tissue, representing 2–10% of the total myonuclei [4]. Moreover, in case of severe or extended damage or in some pathological muscle condition their function may be also compromised and the inefficient muscle regeneration replaces the muscle tissue with a fibrotic not contractile tissue. In such a context, strategies aimed to improve endogenous regenerative potential including SC activity may offer promising perspectives.
Author Contributions
“Conceptualization, F.C., A.T., R.S. and C.S.; methodology, F.C., A.T., R.S. and C.S.; validation, F.C., A.T., M.P., F.P., R.G., R.S. and C.S.; formal analysis, F.C., F.P. and R.S.; investigation, F.C., A.T. M.P., F.P., R.G., R.S., C.S.; resources, F.C., S.Z.-O., R.S, C.S.; data curation, F.C., A.T., F.P., R.S., C.S.; writing—original draft preparation, F.C., A.T., R.S., C.S.; writing—review and editing, F.C., A.T., M.P., F.P., R.G., S.Z.-O., R.S. and C.S.; visualization, F.C., A.T., R.S., C.S.; supervision, C.S.; project administration, R.S., C.S.; funding acquisition, F.C., S.Z.-O., R.S. and C.S. All authors have read and agreed to the published version of the manuscript.”
Funding
“This research was funded by MIUR (Ministry of Education, University and Research, Italy)- Fondi di Ateneo-ex 60%—University of Florence to F.C., S.Z.-O., R.S. and C.S.
Institutional Review Board Statement
“The animal study protocol was conducted according to the guidelines of the Directive 2010/63/EU of the European Parliament and of the European Union council (22 September 2010) on the protection of animals used for scientific purposes and approved by the Italian Ministry of Health (approval code: 787/2016-PR date of approval: 1/09/2016; approval code: 0DD9B.N.ZB6/2020 date of approval: 1/09/2020) and the Institutional Animal Care and Use Committee of the University of Florence, Italy. The ethical policy of the University of Florence observes the Guide for the Care and Use of Laboratory Animals of the US National Institutes of Health (NIH Publication No. 85–23, revised 1996; University of Florence assurance number: A5278-01). The mice were housed at a controlled temperature (21 ± 1 °C) under a 12 h light/12 h dark photoperiod with free access to laboratory food and water. All efforts were made to minimize the animal suffering and the number of animals sacrificed. The animals were killed by a rapid cervical dislocation.”
Data Availability Statement
Data are available from the corresponding author upon reasonable request.
Acknowledgments
The authors acknowledge MIUR-Italy (“Progetto Dipartimenti di Eccellenza 2018−2022”) for the funds allocated to the Department of Experimental and Clinical Medicine.
Conflicts of Interest
“The authors declare no conflict of interest.”
References
- Sousa-Victor, P.; García-Prat, L.; Muñoz-Cánoves, P. Control of satellite cell function in muscle regeneration and its disruption in ageing. Nat. Rev. Mol. Cell Biol. 2022, 23, 204-226. doi: 10.1038/s41580-021-00421-2. Sousa-Victor, P.; García-Prat, L.; Muñoz-Cánoves, P. Control of satellite cell function in muscle regeneration and its disruption in ageing. Nat. Rev. Mol. Cell Biol. 2022, 23, 204-226. [CrossRef] [PubMed]
- Feige, P.; Brun, C.E.; Ritso, M.; Rudnicki, M.A. Orienting Muscle Stem Cells for Regeneration in Homeostasis, Aging, and Disease. Cell Stem Cell 2018, 23, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.L.; Kamal, M.; Parise, G. The Role of Supporting Cell Populations in Satellite Cell Mediated Muscle Repair. Cells 2023, 12, 1968. [Google Scholar] [CrossRef] [PubMed]
- Forcina, L.; Cosentino, M.; Musarò, A. Mechanisms Regulating Muscle Regeneration: Insights into the Interrelated and Time-Dependent Phases of Tissue Healing. Cells 2020, 9, 1297. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Tani, A.; Rosa, I.; Chellini, F.; Squecco, R.; Idrizaj, E.; Zecchi-Orlandini, S.; Ibba-Manneschi, L.; Sassoli, C. Morphological evidence for telocytes as stromal cells supporting satellite cell activation in eccentric contraction-induced skeletal muscle injury. Sci. Rep. 2019, 9, 14515. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.A.; Olsen, I.; Zammit, P.S.; Heslop, L.; Petrie, A.; Partridge, T.A.; Morgan, J.E. Stem Cell Function, Self-Renewal, and Behavioral Heterogeneity of Cells from the Adult Muscle Satellite Cell Niche. Cell 2005, 122, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Takemasa, T.; Fujita, R. High Throughput Screening of Mitochondrial Bioenergetics in Myoblasts and Differentiated Myotubes. Methods Mol. Biol. 2023, 2640, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Singh, V. Intracellular metabolic reprogramming mediated by micro-RNAs in differentiating and proliferating cells under non-diseased conditions. Mol. Biol. Rep. 2021, 48, 8123–8140. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Scimè, A. (2020) Mitochondrial Function in Muscle Stem Cell Fates. Front. Cell Dev. Biol. 2020, 8, 480. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, F.; Mangiavini, L.; La Rocca, P.; Piccoli, M.; Ghiroldi, A.; Rota, P.; Tarantino, A.; Canciani, B.; Coviello, S.; Messina, C.; Ciconte, G.; Pappone, C.; Peretti, G.M.; Anastasia, L. Human Sarcopenic Myoblasts Can Be Rescued by Pharmacological Reactivation of HIF-1α. Int. J. Mol. Sci. 2022, 23, 7114. [Google Scholar] [CrossRef] [PubMed]
- Salekeen, R.; Kyba, M. Not young but still immature: a HIF-1α-mediated maturation checkpoint in regenerating muscle. J. Clin. Invest. 2022, 132, e165322. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Mao, J.; Han, X.; Zhang, W.; Li, Y.; Liu, Y.; Li, Q. Downregulated hypoxia-inducible factor 1α improves myoblast differentiation under hypoxic condition in mouse genioglossus. Mol. Cell. Biochem. 2021, 476, 1351–1364. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Conotte, S.; Belayew, A.; Declèves, A.E.; Legrand, A.; Tassin, A. Hypoxia and Hypoxia-Inducible Factor Signaling in Muscular Dystrophies: Cause and Consequences. Int. J. Mol. Sci. 2021, 22, 7220. [Google Scholar] [CrossRef] [PubMed]
- Pircher, T.; Wackerhage, H.; Aszodi, A.; Kammerlander, C.; Böcker, W.; Saller, M.M. Hypoxic Signaling in Skeletal Muscle Maintenance and Regeneration: A Systematic Review. Front. Physiol. 2021, 12, 684899. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, F.; Resmini, G.; Angelino, E.; Ferrara, M.; Tarantino, A.; Piccoli, M.; Rota, P.; Ghiroldi, A.; Monasky, M.M.; Ciconte, G.; Pappone, C.; Graziani, A. , Anastasia, L. HIF-1α Directly Controls WNT7A Expression During Myogenesis. Front. Cell Dev. Biol. 2020, 8, 593508. [Google Scholar] [CrossRef] [PubMed]
- Sinha, K.M.; Tseng, C.; Guo, P.; Lu, A.; Pan, H.; Gao, X.; Andrews, R.; Eltzschig, H.; Huard, J. Hypoxia-inducible factor 1α (HIF-1α) is a major determinant in the enhanced function of muscle-derived progenitors from MRL/MpJ mice. FASEB J. 2019, 33, 8321–8334. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, F.; Resmini, G.; Ghiroldi, A.; Piccoli, M.; Bergante, S.; Tettamanti, G.; Anastasia, L. Activation of the hypoxia-inducible factor 1α promotes myogenesis through the noncanonical Wnt pathway, leading to hypertrophic myotubes. FASEB J. 2017, 31, 2146–2156. [Google Scholar] [CrossRef]
- Yang, X.; Yang, S.; Wang, C.; Kuang, S. The hypoxia-inducible factors HIF1α and HIF2α are dispensable for embryonic muscle development but essential for postnatal muscle regeneration. J. Biol. Chem. 2017, 292, 5981–5991. [Google Scholar] [CrossRef] [PubMed]
- Dehne, N.; Kerkweg, U.; Otto, T.; Fandrey, J. The HIF-1 response to simulated ischemia in mouse skeletal muscle cells neither enhances glycolysis nor prevents myotube cell death. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1693–1701. [Google Scholar] [CrossRef]
- Li, X.; Zhu, L.; Chen, X.; Fan, M. Effects of hypoxia on proliferation and differentiation of myoblasts. Med. Hypotheses 2007, 69, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Sensui, H.; Sakamoto, Y.; Nagatomi, R. Knockdown of hypoxia-inducible factor-1alpha by siRNA inhibits C2C12 myoblast differentiation. J. Cell. Biochem. 2006, 98, 642–649. [Google Scholar] [CrossRef]
- Kubis, H.P.; Hanke, N.; Scheibe, R.J.; Gros, G. Accumulation and nuclear import of HIF1 alpha during high and low oxygen concentration in skeletal muscle cells in primary culture. Biochim. Biophys. Acta 2005, 1745, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Kim, D.; Rhee, J.; Seo, J.Y.; Park, I.; Kim, J.H.; Lee, D.; Lee, W.; Kim, Y.L.; Yoo, K.; Bae, S.; Chung, J.; Seong, R.H.; Kong, Y.Y. Baf155 regulates skeletal muscle metabolism via HIF-1a signaling. PLoS Biol. 2023, 21, e3002192. [Google Scholar] [CrossRef] [PubMed]
- Hao, T.; Liu, Y.H.; Li, Y.Y.; Lu, Y.; Xu, H.Y. A Transcriptomic Analysis of Physiological Significance of Hypoxia-inducible Factor-1α in Myogenesis and Carbohydrate Metabolism of Genioglossus in Mice. Chin. Med. J. (Engl). 2017, 130, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Kitakaze, T.; Sugihira, T.; Kameyama, H.; Maruchi, A.; Kobayashi, Y.; Harada, N.; Yamaji, R. Carotenoid transporter CD36 expression depends on hypoxia-inducible factor-1α in mouse soleus muscles. J. Clin. Biochem. Nutr. 2022, 71, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Settelmeier, S.; Schreiber, T.; Mäki, J.; Byts, N.; Koivunen, P.; Myllyharju, J.; Fandrey, J.; Winning, S. Prolyl hydroxylase domain 2 reduction enhances skeletal muscle tissue regeneration after soft tissue trauma in mice. PLoS One 2020, 15, e0233261. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Lee, D.S.; Skuli, N.; Mesquita, R.C.; Kim, M.N.; Yodh, A.G.; Nguyen-McCarty, M.; Li, B.; Simon, M.C. HIF modulation of Wnt signaling regulates skeletal myogenesis in vivo. Development. 2015, 142, 2405–2412. [Google Scholar] [CrossRef] [PubMed]
- Cicchillitti, L.; Di Stefano, V.; Isaia, E.; Crimaldi, L.; Fasanaro, P.; Ambrosino, V.; Antonini, A.; Capogrossi, M.C.; Gaetano, C.; Piaggio, G.; Martelli, F. Hypoxia-inducible factor 1-α induces miR-210 in normoxic differentiating myoblasts. J. Biol. Chem. 2012, 287, 44761–44771. [Google Scholar] [CrossRef] [PubMed]
- Wagatsuma, A.; Kotake, N.; Yamada, S. Spatial and temporal expression of hypoxia-inducible factor-1α during myogenesis in vivo and in vitro. Mol. Cell. Biochem. 2011, 347, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Mounier, R.; Pedersen, B.K.; Plomgaard, P. Muscle-specific expression of hypoxia-inducible factor in human skeletal muscle. Exp. Physiol. 2010, 95, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell. 2005, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Stroka, D.M.; Burkhardt, T.; Desbaillets, I.; Wenger, R.H.; Neil, D.A.; Bauer, C.; Gassmann, M.; Candinas, D. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J. 2001, 15, 2445–2453. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Huang, H.; Cui, Y.; Li, W.; Zhang, S.; Chen, Y. Study on the Mechanism of Capillary Leakage Caused by Hypoxia-Inducible Factor-1α through Inducing High Expression of Matrix Metalloproteinase-9. J. Oncol. 2021, 2021, 9130650. [Google Scholar] [CrossRef]
- Li, Y.Y.; Zheng, Y.L. Hypoxia promotes invasion of retinoblastoma cells in vitro by upregulating HIF-1α/MMP9 signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 5361–5369. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Jang, Y.S.; Min, S.Y.; Song, J.Y. Overexpression of MMP-9 and HIF-1α in Breast Cancer Cells under Hypoxic Conditions. J. Breast Cancer 2011, 14, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegué, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; Bergers, G. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Nowak, E.; Gawor, M.; Ciemerych, M.A.; Zimowska, M. Silencing of gelatinase expression delays myoblast differentiation in vitro. Cell. Biol. Int. 2018, 42, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Rebalka, I.A.; Monaco, C.M.F.; Varah, N.E.; Berger, T.; D'souza, D.M.; Zhou, S.; Mak, T.W.; Hawke, T.J. Loss of the adipokine lipocalin-2 impairs satellite cell activation and skeletal muscle regeneration. Am. J. Physiol. Cell Physiol. 2018, 315, C714–C721. [Google Scholar] [CrossRef] [PubMed]
- Sassoli, C.; Nosi, D.; Tani, A.; Chellini, F.; Mazzanti, B.; Quercioli, F.; Zecchi-Orlandini, S.; Formigli, L. Defining the role of mesenchymal stromal cells on the regulation of matrix metalloproteinases in skeletal muscle cells. Exp. Cell Res. 2014, 323, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Zimowska, M.; Swierczynska, M.; Ciemerych, M.A. Nuclear MMP-9 role in the regulation of rat skeletal myoblasts proliferation. Biol. Cell. 2013, 105, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Squecco, R.; Chellini, F.; Idrizaj, E.; Tani, A.; Garella, R.; Pancani, S.; Pavan, P.; Bambi, F.; Zecchi-Orlandini, S.; Sassoli, C. Platelet-Rich Plasma Modulates Gap Junction Functionality and Connexin 43 and 26 Expression During TGF-β1-Induced Fibroblast to Myofibroblast Transition: Clues for Counteracting Fibrosis. Cells 2020, 9, 1199. [Google Scholar] [CrossRef] [PubMed]
- Sassoli, C.; Vallone, L.; Tani, A.; Chellini, F.; Nosi, D.; Zecchi-Orlandini, S. Combined use of bone marrow-derived mesenchymal stromal cells (BM-MSCs) and platelet rich plasma (PRP) stimulates proliferation and differentiation of myoblasts in vitro: new therapeutic perspectives for skeletal muscle repair/regeneration. Cell Tissue Res. 2018, 372, 549–570. [Google Scholar] [CrossRef] [PubMed]
- Bernacchioni, C.; Ghini, V.; Squecco, R.; Idrizaj, E.; Garella, R.; Puliti, E.; Cencetti, F.; Bruni, P.; Donati, C. Role of Sphingosine 1-Phosphate Signalling Axis in Muscle Atrophy Induced by TNFα in C2C12 Myotubes. Int. J. Mol. Sci. 2021, 22, 1280. [Google Scholar] [CrossRef] [PubMed]
- Formigli, L.; Sassoli, C.; Squecco, R.; Bini, F.; Martinesi, M.; Chellini, F.; Luciani, G.; Sbrana, F.; Zecchi-Orlandini, S.; Francini, F.; Meacci, E. Regulation of transient receptor potential canonical channel 1 (TRPC1) by sphingosine 1-phosphate in C2C12 myoblasts and its relevance for a role of mechanotransduction in skeletal muscle differentiation. J. Cell Sci. 2009, 122, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Martella, D.; Mannelli, M.; Squecco, R.; Garella, R.; Idrizaj, E.; Antonioli, D.; Laus, M.; Wiersma, D.S.; Gamberi, T.; Paoli, P.; Parmeggiani, C.; Fiaschi, T. Cell instructive Liquid Crystalline Networks for myotube formation. iScience 2021, 24, 103077. [Google Scholar] [PubMed]
- Sassoli, C.; Pini, A.; Chellini, F.; Mazzanti, B.; Nistri, S.; Nosi, D.; Saccardi, R.; Quercioli, F.; Zecchi-Orlandini, S.; Formigli, L. Bone marrow mesenchymal stromal cells stimulate skeletal myoblast proliferation through the paracrine release of VEGF. PLoS One 2012, 7, e37512. [Google Scholar] [CrossRef]
- Luo, D.; Renault, V.M.; Rando, T.A. The regulation of Notch signaling in muscle stem cell activation and postnatal myogenesis. Semin. Cell Dev. Biol. 2005, 16, 612–622. [Google Scholar] [CrossRef]
- Formigli, L.; Meacci, E.; Sassoli, C.; Squecco, R.; Nosi, D.; Chellini, F.; Naro, F.; Francini, F.; Zecchi-Orlandini, S. (2007). Cytoskeleton/stretch-activated ion channel interaction regulates myogenic differentiation of skeletal myoblasts. J. Cell. Physiol. 2007, 211, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Sin, J.; Andres, A.M.; Taylor, D.J.; Weston, T.; Hiraumi, Y.; Stotland, A.; Kim, B.J.; Huang, C.; Doran, K.S.; Gottlieb, R.A. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 2016, 12, 369–380. [Google Scholar] [CrossRef]
- Wagatsuma, A.; Sakuma, K. Mitochondria as a potential regulator of myogenesis. ScientificWorldJournal 2013, 2013, 593267. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiol. (Bethesda) 2009, 24, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Summermatter, S.; Santos, G.; Pérez-Schindler, J.; Handschin, C. Skeletal muscle PGC-1α controls whole-body lactate homeostasis through estrogen-related receptor α-dependent activation of LDH B and repression of LDH A. Proc. Natl. Acad. Sci. U S A. 2013, 110, 8738–8743. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef]
- Ling, M.; Quan, L.; Lai, X.; Lang, L.; Li, F.; Yang, X.; Fu, Y.; Feng, S.; Yi, X.; Zhu, C.; Gao, P.; Zhu, X.; Wang, L.; Shu, G.; Jiang, Q.; Wang, S. VEGFB Promotes Myoblasts Proliferation and Differentiation through VEGFR1-PI3K/Akt Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 13352. [Google Scholar] [CrossRef] [PubMed]
- Arsic, N.; Zacchigna, S.; Zentilin, L.; Ramirez-Correa, G.; Pattarini, L.; Salvi, A.; Sinagra, G.; Giacca, M. Vascular endothelial growth factor stimulates skeletal muscle regeneration in vivo. Mol. Ther. 2004, 10, 844–854. [Google Scholar] [CrossRef]
- Germani, A.; Di Carlo, A.; Mangoni, A.; Straino, S.; Giacinti, C.; Turrini, P.; Biglioli, P.; Capogrossi, M.C. Vascular endothelial growth factor modulates skeletal myoblast function. Am. J. Pathol. 2003, 163, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Basic, V.T.; Jacobsen, A.; Sirsjö, A.; Abdel-Halim, S.M. TNF stimulation induces VHL overexpression and impairs angiogenic potential in skeletal muscle myocytes. Int. J. Mol. Med. 2014, 34, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, R.P.; Johnson, R.M.; Rathbone, C.R.; Liu, X.; Temm-Grove, C.; Sheehan, S.M.; Hoying, J.B.; Allen, R.E. Satellite cell-mediated angiogenesis in vitro coincides with a functional hypoxia-inducible factor pathway. Am. J. Physiol. Cell Physiol. 2009, 296, C1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Semenza, G.L. Regulation of angiogenesis by hypoxia-inducible factor 1. Crit. Rev. Oncol. Hematol. 2006, 59, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Olson, N.; van der Vliet, A. Interactions between nitric oxide and hypoxia-inducible factor signaling pathways in inflammatory disease. Nitric Oxide. 2011, 25, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Déry, M.A.; Michaud, M.D.; Richard, D.E. Hypoxia-inducible factor 1: regulation by hypoxic and non-hypoxic activators. Int. J. Biochem. Cell Biol. 2005, 37, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Sibisi, N.C.; Snyman, C.; Myburgh, K.H.; Niesler, C.U. Evaluating the role of nitric oxide in myogenesis in vitro. Biochimie. 2022, 196, 216–224. [Google Scholar] [CrossRef] [PubMed]
- O'Hagan, K.A.; Cocchiglia, S.; Zhdanov, A.V.; Tambuwala, M.M.; Cummins, E.P.; Monfared, M.; Agbor, T.A.; Garvey, J.F.; Papkovsky, D.B.; Taylor, C.T.; Allan, B.B. PGC-1alpha is coupled to HIF-1alpha-dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proc. Natl. Acad. Sci. U S A. 2009, 106, 2188–2193. [Google Scholar] [CrossRef] [PubMed]
- Villa, J.C.; Chiu, D.; Brandes, A.H.; Escorcia, F.E.; Villa, C.H.; Maguire, W.F.; Hu, C.J.; de Stanchina, E.; Simon, M.C.; Sisodia, S.S.; Scheinberg, D.A.; and Li, Y.M. Nontranscriptional role of Hif-1 α in activation of γ-secretase and notch signaling in breast cancer. Cell Rep. 2014, 8, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Sakagami, H.; Makino, Y.; Mizumoto, K.; Isoe, T.; Takeda, Y.; Watanabe, J.; Fujita, Y.; Takiyama, Y.; Abiko, A.; Haneda, M. Loss of HIF-1α impairs GLUT4 translocation and glucose uptake by the skeletal muscle cells. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1065–E1076. [Google Scholar] [CrossRef] [PubMed]
- Hubbi, M.E.; Kshitiz; Gilkes, D.M.; Rey, S.; Wong, C.C.; Luo, W.; Kim, D.H.; Dang, C.V.; Levchenko, A.; Semenza, G.L. A nontranscriptional role for HIF-1α as a direct inhibitor of DNA replication. Sci. Signal. 2013, 6, ra10. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Schüler, S.C.; Hüttner, S.S.; von Eyss, B.; von Maltzahn, J. Adult stem cells at work: regenerating skeletal muscle. Cell. Mol. Life Sci. 2019, 76, 2559–2570. [Google Scholar] [CrossRef] [PubMed]
- Yun, Z.; Lin, Q.; Giaccia, A.J. Adaptive myogenesis under hypoxia. Mol. Cell. Biol. 2005, 25, 3040–3055. [Google Scholar] [CrossRef]
- Zimowska, M.; Olszynski, K.H.; Swierczynska, M.; Streminska, W.; Ciemerych, M.A. Decrease of MMP-9 activity improves soleus muscle regeneration. Tissue Eng. Part A. 2012, 18, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Kann, A.P.; Hung, M.; Wang, W.; Nguyen, J.; Gilbert, P.M.; Wu, Z.; Krauss, R.S. An injury-responsive Rac-to-Rho GTPase switch drives activation of muscle stem cells through rapid cytoskeletal remodeling. Cell Stem Cell. 2022, 29, 933–947.e6. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Z.G.; Taiyab, A.; West-Mays, J.A. MMP9 Differentially Regulates Proteins Involved in Actin Polymerization and Cell Migration during TGF-β-Induced EMT in the Lens. Int. J. Mol. Sci. 2021, 22, 11988. [Google Scholar] [CrossRef] [PubMed]
- Bernacchioni, C.; Squecco, R.; Gamberi, T.; Ghini, V.; Schumacher, F.; Mannelli, M.; Garella, R.; Idrizaj, E.; Cencetti, F.; Puliti, E.; Bruni, P.; Turano, P.; Fiaschi, T.; Donati, C. S1P Signalling Axis Is Necessary for Adiponectin-Directed Regulation of Electrophysiological Properties and Oxidative Metabolism in C2C12 Myotubes. Cells, 2022, 11, 713. [Google Scholar] [CrossRef] [PubMed]
- Muratore, M.; Srsen, V.; Waterfall, M.; Downes, A.; Pethig, R. Biomarker-free dielectrophoretic sorting of differentiating myoblast multipotent progenitor cells and their membrane analysis by Raman spectroscopy. Biomicrofluidics. 2012, 6, 34113. [Google Scholar] [CrossRef] [PubMed]
- Meacci, E.; Bini, F.; Sassoli, C.; Martinesi, M.; Squecco, R.; Chellini, F.; Zecchi-Orlandini, S.; Francini, F.; Formigli, L. Functional interaction between TRPC1 channel and connexin-43 protein: a novel pathway underlying S1P action on skeletal myogenesis. Cell. Mol. Life Sci. 2010, 67, 4269–4285. [Google Scholar] [CrossRef] [PubMed]
- Shimahara, T.; Bournaud, R. Barium currents in developing skeletal muscle cells of normal and mutant mice foetuses with 'muscular dysgenesis'. Cell Calcium. 1991, 12, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Tamayo, T.; Grajales, L.; García, J. Commitment of satellite cells expressing the calcium channel α2δ1 subunit to the muscle lineage. J. Signal Transduct. 2012, 2012, 460842. [Google Scholar] [CrossRef] [PubMed]
- Grajales, L.; Lach, L.E.; Janisch, P.; Geenen, D.L.; García, J. Temporal Expression of Calcium Channel Subunits in Satellite Cells and Bone Marrow Mesenchymal Cells. Stem Cell Rev. Rep. 2015, 11, 408–422. [Google Scholar] [CrossRef] [PubMed]
- García, K.; Nabhani, T.; & García, J. The calcium channel α2/δ1 subunit is involved in extracellular signaling. J. Physiol. 2008, 586, 727–738. [CrossRef] [PubMed]
- Wang, J.; Weigand, L.; Lu, W.; Sylvester, J.T.; Semenza, G.L.; Shimoda, L.A. Hypoxia Inducible Factor 1 Mediates Hypoxia-Induced TRPC Expression and Elevated Intracellular Ca2+ in Pulmonary Arterial Smooth Muscle Cells. Circ. Res. 2006, 98, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Morphological analysis of C2C12 myoblast differentiation under normoxia. C2C12 cells were cultured in the proliferation medium (PM) for 24h or in myogenic differentiation medium (DM) for different times (24, 48, 72h and 5d). (A-L) Representative confocal immunofluorescence images of C2C12 cells immunostained with antibodies against (A-D) the nuclear protein Ki67 (green), (E-G) MyoD (green), (H) Myogenin (red) and (I-L) Notch-1 (green). (M,N) Quantitative analyses of Ki67 and MyoD positive nuclei expressed as the percentage of the total nuclei, respectively. (O) Densitometric analysis of Notch-1 fluorescence signal intensity (F.I.) in the indicated experimental condition, performed on digitized images in 10 regions of interest (ROI) of 100 μm2 for each confocal stack (10). a.u.: arbitrary units. (P) Representative phase contrast light microscopic image of myotube formation after 5d culture in DM. (Q,R) Representative confocal immunofluorescence images of C2C12 cells grown in DM for 5d immunostained with antibodies against myogenin (red) and sarcomeric actin (green) respectively. In (Q) the cells were counterstained with-phalloidin to visualize F-actin filaments (green). Scale bar: in (A-L), (Q) and (R), 25 µm; in (P), 200 µm. Data are reported as mean ± SD of at least three independent experiments performed in triplicates. Significance of difference: * p<0.05, ** p<0.01 vs PM; °° p<0.01 vs DM 24h; # p< 0.05, ## p<0.01 vs DM 48h (One-way ANOVA with post-hoc Tukey HSD).
Figure 1.
Morphological analysis of C2C12 myoblast differentiation under normoxia. C2C12 cells were cultured in the proliferation medium (PM) for 24h or in myogenic differentiation medium (DM) for different times (24, 48, 72h and 5d). (A-L) Representative confocal immunofluorescence images of C2C12 cells immunostained with antibodies against (A-D) the nuclear protein Ki67 (green), (E-G) MyoD (green), (H) Myogenin (red) and (I-L) Notch-1 (green). (M,N) Quantitative analyses of Ki67 and MyoD positive nuclei expressed as the percentage of the total nuclei, respectively. (O) Densitometric analysis of Notch-1 fluorescence signal intensity (F.I.) in the indicated experimental condition, performed on digitized images in 10 regions of interest (ROI) of 100 μm2 for each confocal stack (10). a.u.: arbitrary units. (P) Representative phase contrast light microscopic image of myotube formation after 5d culture in DM. (Q,R) Representative confocal immunofluorescence images of C2C12 cells grown in DM for 5d immunostained with antibodies against myogenin (red) and sarcomeric actin (green) respectively. In (Q) the cells were counterstained with-phalloidin to visualize F-actin filaments (green). Scale bar: in (A-L), (Q) and (R), 25 µm; in (P), 200 µm. Data are reported as mean ± SD of at least three independent experiments performed in triplicates. Significance of difference: * p<0.05, ** p<0.01 vs PM; °° p<0.01 vs DM 24h; # p< 0.05, ## p<0.01 vs DM 48h (One-way ANOVA with post-hoc Tukey HSD).
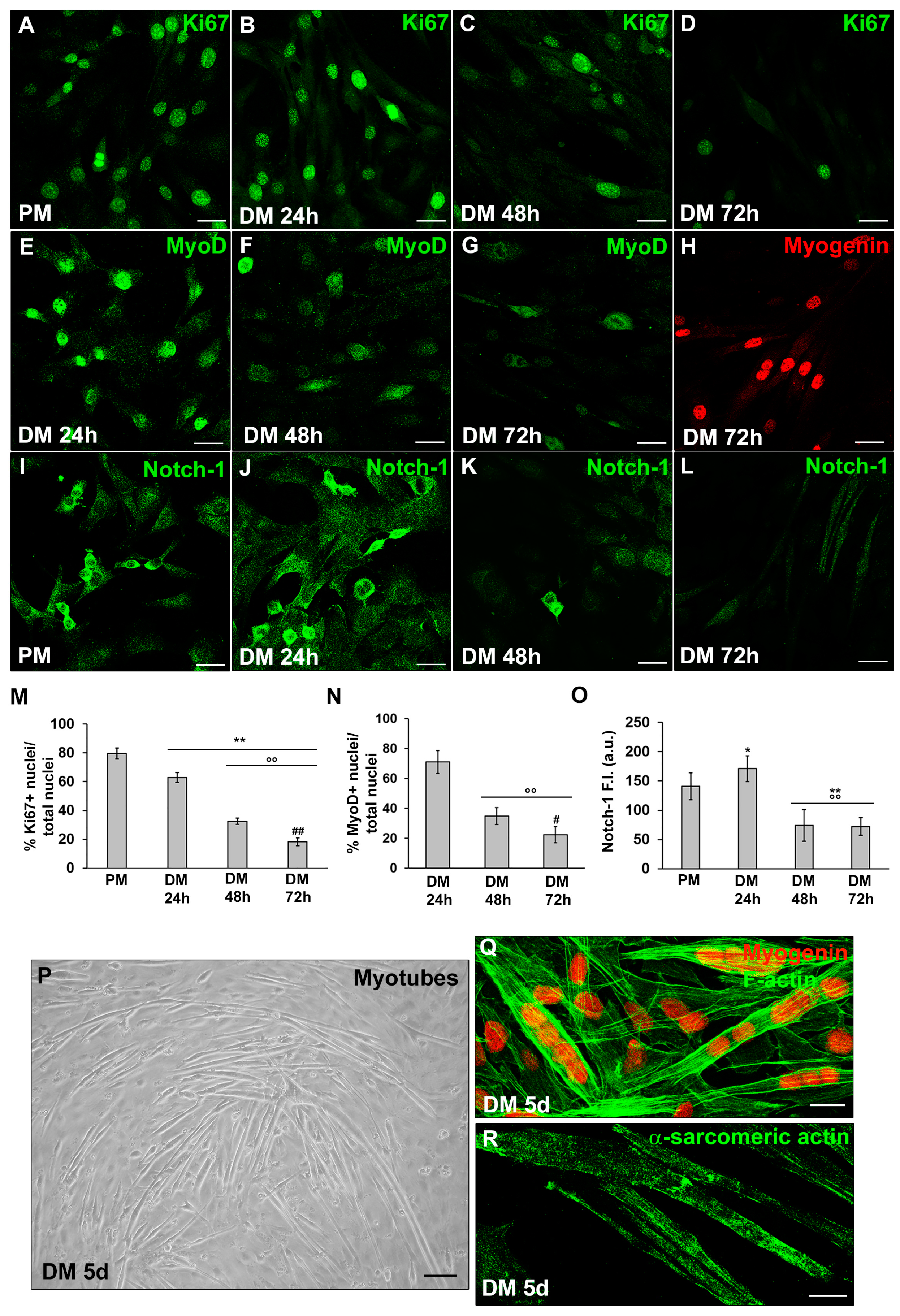
Figure 2.
Evaluation of the SDH-B, LDH-A and PGC-1α during C2C12 myoblast differentiation. The cells were cultured in proliferation medium (PM) for 24h or in myogenic differentiation medium (DM) for different times (24, 48, 72h and 5d). (A,B) Western Blotting analysis of SDH-B, LDH-A: representative blot and bar charts showing the densitometric analysis of the bands normalized to α-tubulin. (C–G) Representative confocal fluorescence images of the cells immunostained with antibodies against PGC-1α (green). Scale bar: 25 µm. (H) Bar charts showing the densitometric analysis of the PGC-1α fluorescent signal intensity (F.I.) performed on digitized images in 10 regions of interest (ROI) of 100 μm2 for each confocal stack (10). a.u.: arbitrary units. Data shown are mean ± SD and represent the results of at least three independent experiments performed in triplicate. Significance of difference: * p<0.05, ** p<0.01 vs PM; °° p<0.01 vs DM 24h; # p<0.05, ## p<0.01 vs DM 48h (One-way ANOVA with post-hoc Tukey HSD).
Figure 2.
Evaluation of the SDH-B, LDH-A and PGC-1α during C2C12 myoblast differentiation. The cells were cultured in proliferation medium (PM) for 24h or in myogenic differentiation medium (DM) for different times (24, 48, 72h and 5d). (A,B) Western Blotting analysis of SDH-B, LDH-A: representative blot and bar charts showing the densitometric analysis of the bands normalized to α-tubulin. (C–G) Representative confocal fluorescence images of the cells immunostained with antibodies against PGC-1α (green). Scale bar: 25 µm. (H) Bar charts showing the densitometric analysis of the PGC-1α fluorescent signal intensity (F.I.) performed on digitized images in 10 regions of interest (ROI) of 100 μm2 for each confocal stack (10). a.u.: arbitrary units. Data shown are mean ± SD and represent the results of at least three independent experiments performed in triplicate. Significance of difference: * p<0.05, ** p<0.01 vs PM; °° p<0.01 vs DM 24h; # p<0.05, ## p<0.01 vs DM 48h (One-way ANOVA with post-hoc Tukey HSD).
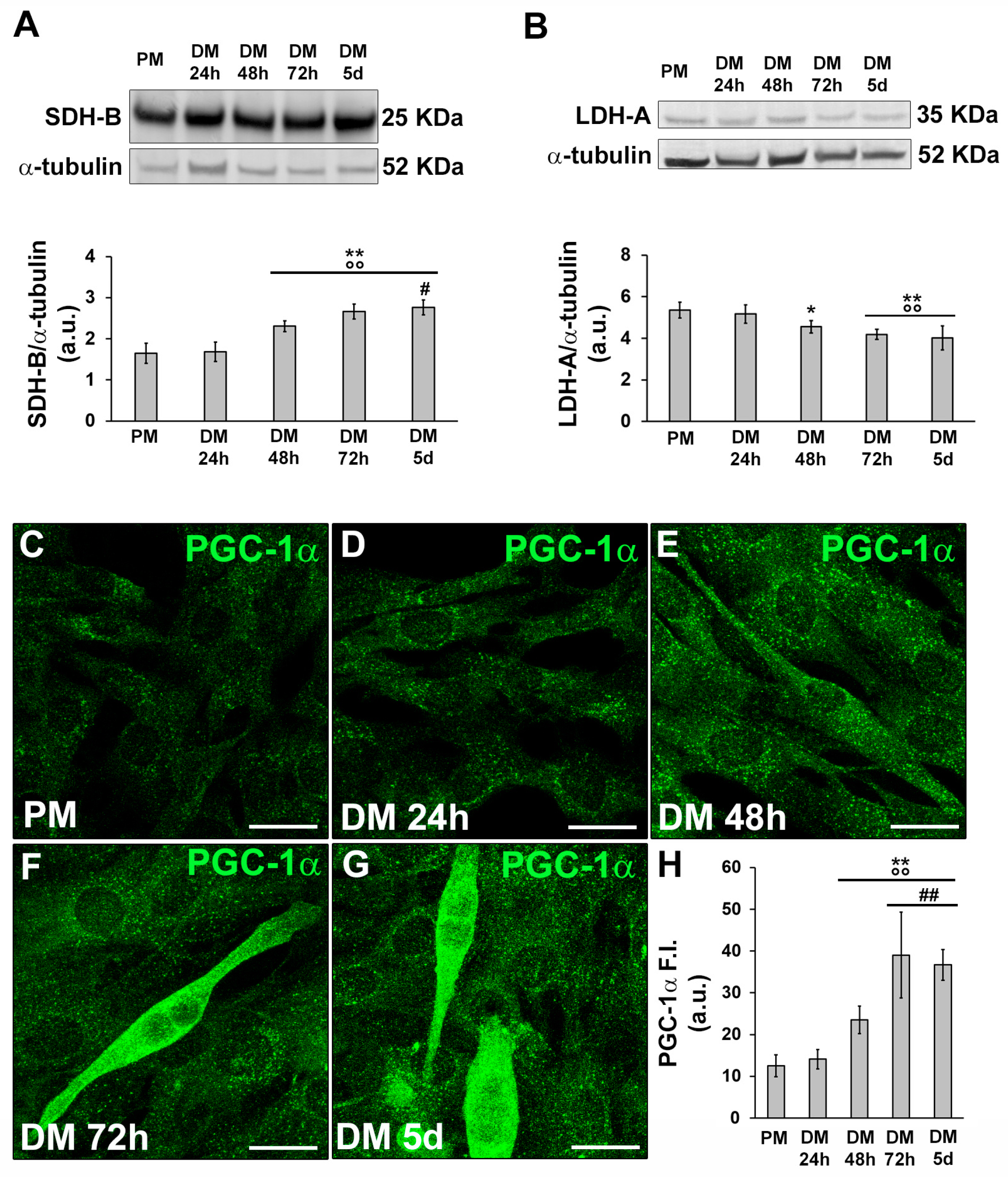
Figure 3.
Evaluation of HIF-1α expression and cellular localization during C2C12 myoblast differentiation. The cells were cultured in proliferation medium (PM) for 24h or in myogenic differentiation medium (DM) for different times (24, 48, 72h and 5d). (A) RT-PCR analysis. Representative agarose gel and bar charts showing the densitometric analysis of the bands normalized to β-actin. a.u.: arbitrary units. (B) Western Blotting analysis. Representative blot and bar charts showing the densitometric analysis of the bands normalized to α-tubulin. Data shown are mean ± SD and represent the results of at least three independent experiments performed in triplicate. (C) Representative confocal fluorescence images of the cells immunostained with antibodies against HIF-1α (green). Note the nuclear localization of the factor in the early differentiating cells. Scale bar: 25 µm. Significance of difference: ** p<0.01 vs PM; °° p<0.01 vs DM 24h; ## p<0.01 vs DM 48h (One-way ANOVA with post-hoc Tukey HSD).
Figure 3.
Evaluation of HIF-1α expression and cellular localization during C2C12 myoblast differentiation. The cells were cultured in proliferation medium (PM) for 24h or in myogenic differentiation medium (DM) for different times (24, 48, 72h and 5d). (A) RT-PCR analysis. Representative agarose gel and bar charts showing the densitometric analysis of the bands normalized to β-actin. a.u.: arbitrary units. (B) Western Blotting analysis. Representative blot and bar charts showing the densitometric analysis of the bands normalized to α-tubulin. Data shown are mean ± SD and represent the results of at least three independent experiments performed in triplicate. (C) Representative confocal fluorescence images of the cells immunostained with antibodies against HIF-1α (green). Note the nuclear localization of the factor in the early differentiating cells. Scale bar: 25 µm. Significance of difference: ** p<0.01 vs PM; °° p<0.01 vs DM 24h; ## p<0.01 vs DM 48h (One-way ANOVA with post-hoc Tukey HSD).
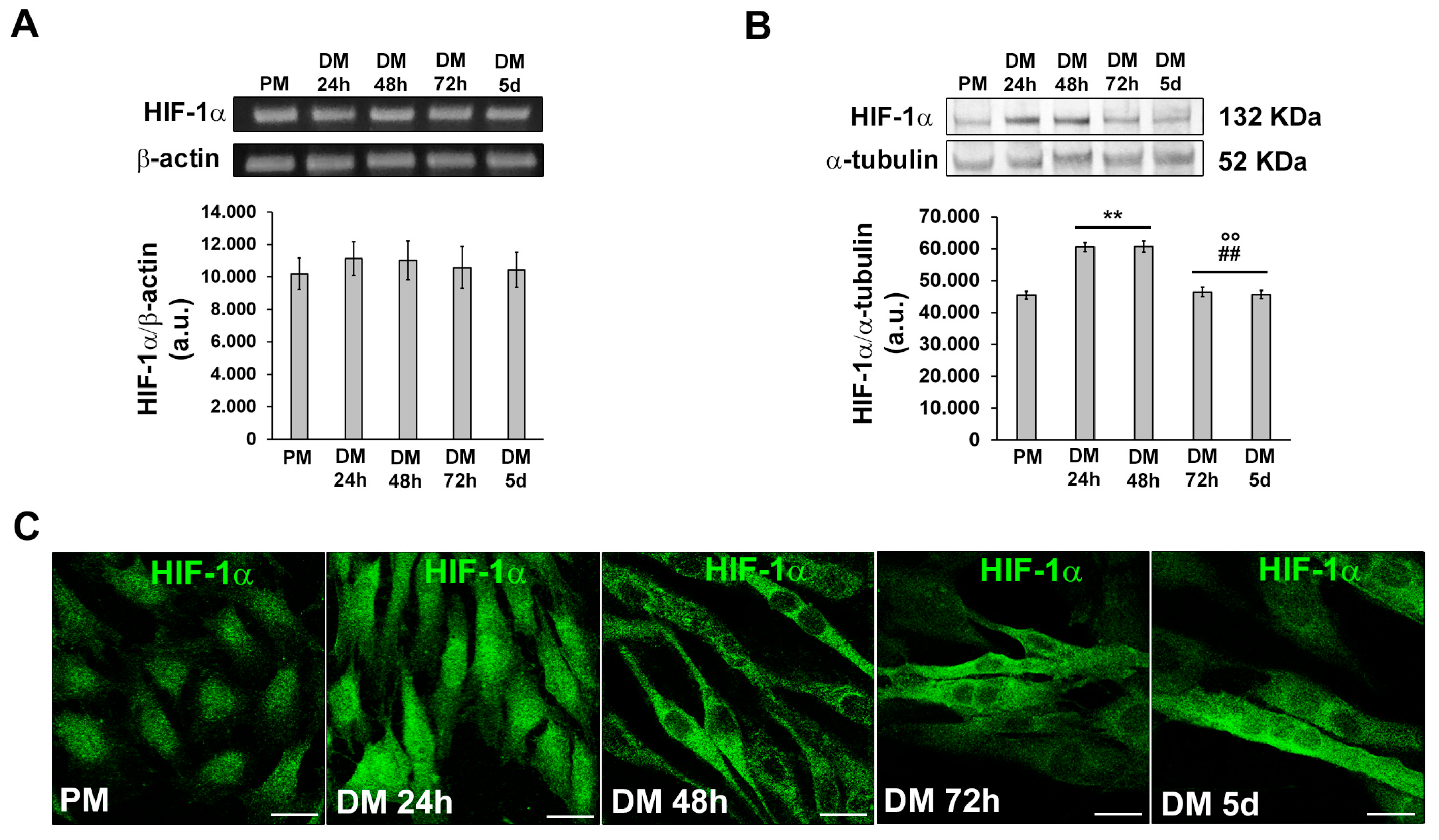
Figure 4.
Evaluation of MMP-9 expression and cellular localization during C2C12 myoblast differentiation. The cells were cultured in proliferation medium (PM) for 24h or in myogenic differentiation medium (DM) for different times (24, 48, 72h and 5d). (A) RT-PCR analysis. Representative agarose gel and bar charts showing the densitometric analysis of the bands normalized to β-actin. a.u.: arbitrary units. (B) Western Blotting analysis. Representative blot and bar charts showing the densitometric analysis of the bands normalized to α-tubulin. (C-G) Representative confocal fluorescence images of the cells immunostained with antibodies against MMP-9 (green). Scale bar: 25 µm. (H) Bar charts showing the densitometric analysis of the MMP-9 fluorescent signal intensity (F.I.) performed on digitized images in 10 regions of interest (ROI) of 100 μm2 for each confocal stack (10). a.u.: arbitrary units. Data shown are mean ± SD and represent the results of at least three independent experiments performed in triplicate. Significance of difference: ** p<0.01 vs PM; °° p<0.01 vs DM 24h; # p<0.05, ## p<0.01 vs DM 48h (One-way ANOVA with post-hoc Tukey HSD).
Figure 4.
Evaluation of MMP-9 expression and cellular localization during C2C12 myoblast differentiation. The cells were cultured in proliferation medium (PM) for 24h or in myogenic differentiation medium (DM) for different times (24, 48, 72h and 5d). (A) RT-PCR analysis. Representative agarose gel and bar charts showing the densitometric analysis of the bands normalized to β-actin. a.u.: arbitrary units. (B) Western Blotting analysis. Representative blot and bar charts showing the densitometric analysis of the bands normalized to α-tubulin. (C-G) Representative confocal fluorescence images of the cells immunostained with antibodies against MMP-9 (green). Scale bar: 25 µm. (H) Bar charts showing the densitometric analysis of the MMP-9 fluorescent signal intensity (F.I.) performed on digitized images in 10 regions of interest (ROI) of 100 μm2 for each confocal stack (10). a.u.: arbitrary units. Data shown are mean ± SD and represent the results of at least three independent experiments performed in triplicate. Significance of difference: ** p<0.01 vs PM; °° p<0.01 vs DM 24h; # p<0.05, ## p<0.01 vs DM 48h (One-way ANOVA with post-hoc Tukey HSD).

Figure 5.
HIF-1α and MMP-9 expression in satellite cells (SCs). A murine SC-enriched culture was obtained from single skeletal EDL muscle fiber as reported in Material and methods. The cells were cultured for 24 h and 48 h in their specific SC proliferation medium. Representative immunofluorescence confocal images of fixed SCs in the indicated experimental conditions, immunostained with (A,B) antibodies against Pax7 (red) and HIF-1α (green), (C,D) antibodies against Pax7 (red) and MMP-9 (green).
Figure 5.
HIF-1α and MMP-9 expression in satellite cells (SCs). A murine SC-enriched culture was obtained from single skeletal EDL muscle fiber as reported in Material and methods. The cells were cultured for 24 h and 48 h in their specific SC proliferation medium. Representative immunofluorescence confocal images of fixed SCs in the indicated experimental conditions, immunostained with (A,B) antibodies against Pax7 (red) and HIF-1α (green), (C,D) antibodies against Pax7 (red) and MMP-9 (green).
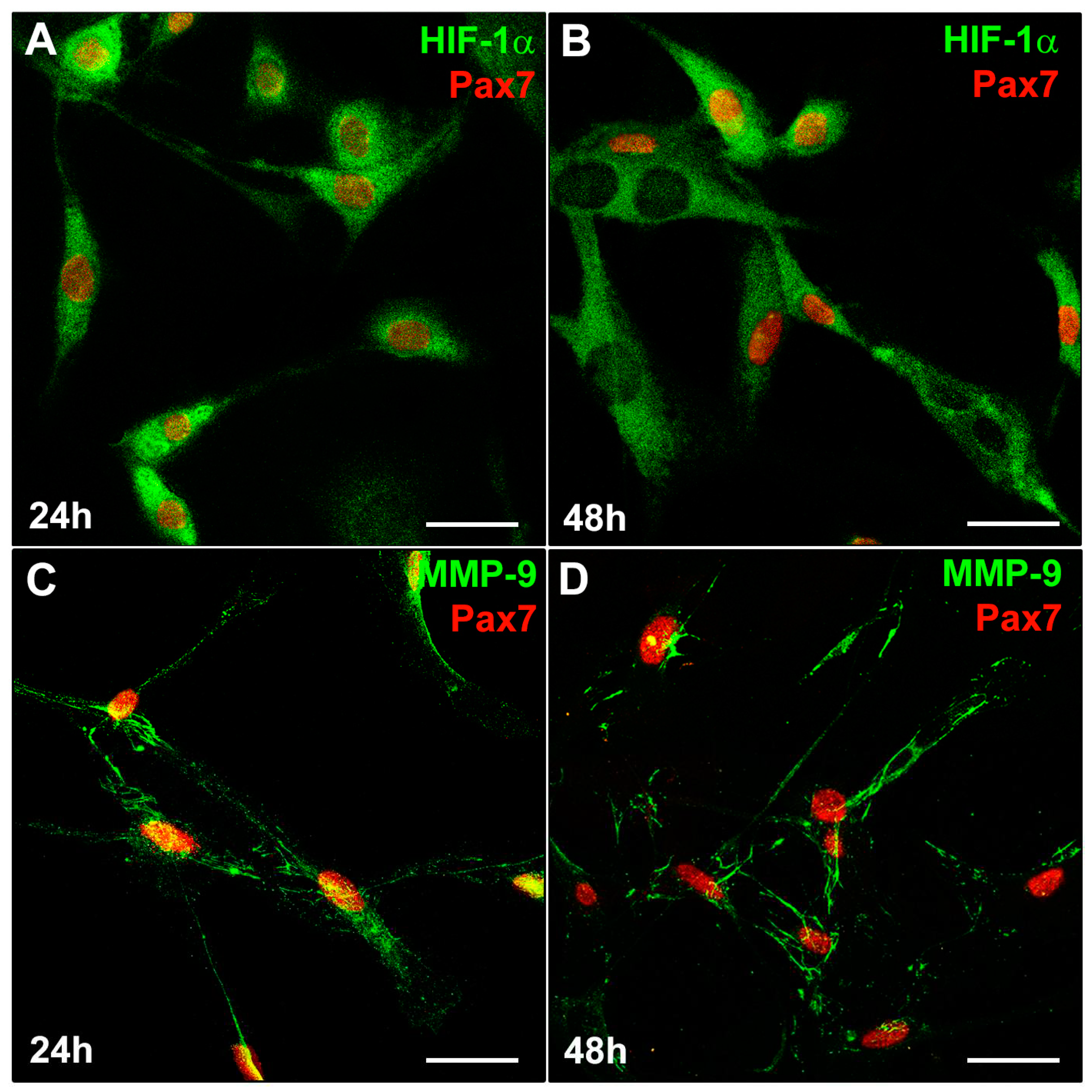
Figure 6.
Evaluation of HIF-1α silencing on C2C12 myoblast MyoD, Notch-ICD and MMP-9 expression and morphology. The cells were transfected in the proliferation medium (PM) with either scrambled siRNA (SCR-siRNA) or HIF-1α-siRNA. After transfection -PM (T0)- the cells were induced to differentiate by shifting in the differentiation medium (DM) for 24 h. WT: wild type not transfected cells. (A,B) Western Blotting analysis. Representative blot and bar charts showing the densitometric analysis of the bands normalized to α-tubulin. (C) Representative confocal fluorescence images of the cells immunostained with antibodies against HIF-1α (green). Scale bar: 25 µm. Bar charts showing the densitometric analysis of the HIF-1α fluorescent signal intensity (F.I.) performed on digitized images in 10 regions of interest (ROI) of 100 μm2 for each confocal stack (10). a.u.: arbitrary units. Data shown are mean ± SD and represent the results of at least three independent experiments performed in triplicate. Significance of difference: ** p<0.01 vs WT PM (T0); ° p<0.05, °° p<0.01 vs SCR-siRNA PM (T0); ## p<0.01 vs WT DM 24h; §§ p<0.01 vs SCR-siRNA DM 24h (One-way ANOVA with post-hoc Tukey HSD). (D) Representative differential interference contrast (DIC) images of fixed C2C12 cells in the indicated experimental conditions.
Figure 6.
Evaluation of HIF-1α silencing on C2C12 myoblast MyoD, Notch-ICD and MMP-9 expression and morphology. The cells were transfected in the proliferation medium (PM) with either scrambled siRNA (SCR-siRNA) or HIF-1α-siRNA. After transfection -PM (T0)- the cells were induced to differentiate by shifting in the differentiation medium (DM) for 24 h. WT: wild type not transfected cells. (A,B) Western Blotting analysis. Representative blot and bar charts showing the densitometric analysis of the bands normalized to α-tubulin. (C) Representative confocal fluorescence images of the cells immunostained with antibodies against HIF-1α (green). Scale bar: 25 µm. Bar charts showing the densitometric analysis of the HIF-1α fluorescent signal intensity (F.I.) performed on digitized images in 10 regions of interest (ROI) of 100 μm2 for each confocal stack (10). a.u.: arbitrary units. Data shown are mean ± SD and represent the results of at least three independent experiments performed in triplicate. Significance of difference: ** p<0.01 vs WT PM (T0); ° p<0.05, °° p<0.01 vs SCR-siRNA PM (T0); ## p<0.01 vs WT DM 24h; §§ p<0.01 vs SCR-siRNA DM 24h (One-way ANOVA with post-hoc Tukey HSD). (D) Representative differential interference contrast (DIC) images of fixed C2C12 cells in the indicated experimental conditions.
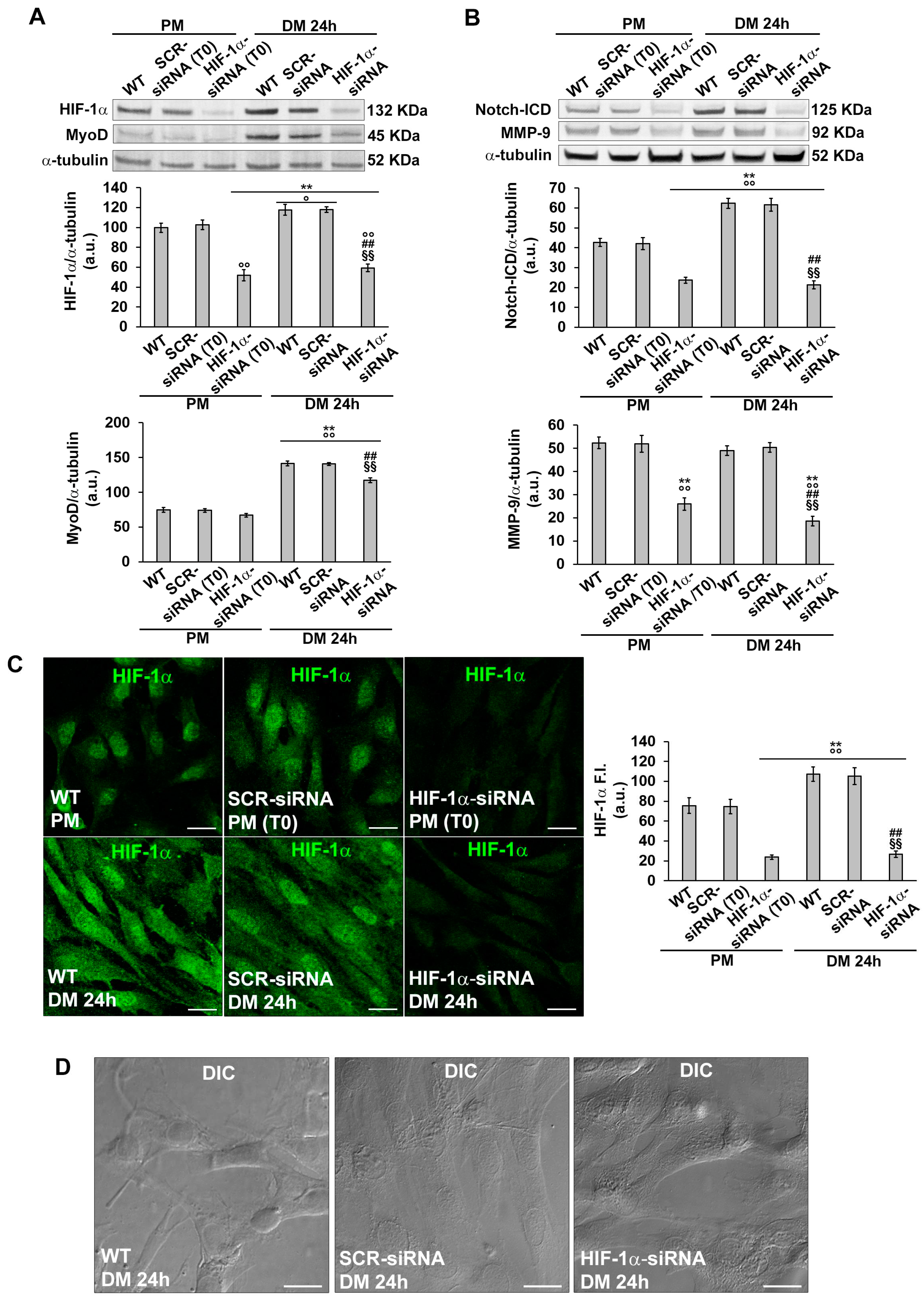
Figure 7.
Evaluation of HIF-1α silencing on C2C12 myoblast Ki67, MyoD, Notch-1 and MMP-9 expression. The cells were transfected with either scrambled siRNA (SCR-siRNA) or HIF-1α-siRNA in PM (T0). After transfection the cells were induced to differentiate by shifting in the differentiation medium (DM) for 24 h. WT: wild type not transfected cells. (A-C,E-GI-K,M-O) Representative confocal fluorescence images of the cells immunostained with antibodies against the indicated markers (green). Scale bar: 25 µm. (D,H) Quantitative analyses of Ki67 and MyoD positive nuclei expressed as the percentage of the total nuclei. (L,P) Bar charts showing densitometric analysis of the Notch-1 and MMP-9 fluorescent signal intensity (F.I.) performed on digitized images in 10 regions of interest (ROI) of 100 μm2 for each confocal stack (10). a.u.: arbitrary units. Data shown are mean ± SD and represent the results of at least three independent experiments performed in triplicate. Significance of difference: ** p<0.01 vs WT DM 24h; °° p<0.01 vs SCR-siRNA DM 24h (One-way ANOVA with post-hoc Tukey HSD).
Figure 7.
Evaluation of HIF-1α silencing on C2C12 myoblast Ki67, MyoD, Notch-1 and MMP-9 expression. The cells were transfected with either scrambled siRNA (SCR-siRNA) or HIF-1α-siRNA in PM (T0). After transfection the cells were induced to differentiate by shifting in the differentiation medium (DM) for 24 h. WT: wild type not transfected cells. (A-C,E-GI-K,M-O) Representative confocal fluorescence images of the cells immunostained with antibodies against the indicated markers (green). Scale bar: 25 µm. (D,H) Quantitative analyses of Ki67 and MyoD positive nuclei expressed as the percentage of the total nuclei. (L,P) Bar charts showing densitometric analysis of the Notch-1 and MMP-9 fluorescent signal intensity (F.I.) performed on digitized images in 10 regions of interest (ROI) of 100 μm2 for each confocal stack (10). a.u.: arbitrary units. Data shown are mean ± SD and represent the results of at least three independent experiments performed in triplicate. Significance of difference: ** p<0.01 vs WT DM 24h; °° p<0.01 vs SCR-siRNA DM 24h (One-way ANOVA with post-hoc Tukey HSD).

Figure 8.
Evaluation of MMP-9 silencing or inhibition on C2C12 myoblast MyoD, Notch-1 and Ki67 expression and morphology. The cells were transfected in the proliferation medium (PM) with either scrambled siRNA (SCR-siRNA) or MMP-9-siRNA. After transfection - PM (T0) - the cells were induced to differentiate by shifting in the differentiation medium (DM) for 24 h. In some experiments the cells were cultured in DM in the presence of the MMP-9 inhibitor SB-3CT (10 µM). WT: wild type not-transfected or untreated cells. (A) Western Blotting analysis. Representative blot and bar charts showing the densitometric analysis of the bands normalized to α-tubulin. (B-E) Representative differential interference contrast (DIC) images of fixed C2C12 cells in the indicated experimental conditions. Scale bar: 12.5 µm. (F-H,J-U) Representative confocal fluorescence images of the cells immunostained with antibodies against the indicated markers (green). Scale bar: 25 µm. (I,X) Bar charts showing the densitometric analysis of the MMP-9 and Notch-1 fluorescent signal intensity (F.I.) performed on digitized images in 10 regions of interest (ROI) of 100 μm2 for each confocal stack (10). a.u.: arbitrary units. (V,W) Quantitative analyses of Ki67 and MyoD positive nuclei expressed as the percentage of the total nuclei, respectively. Data shown are mean ± SD and represent the results of at least three independent experiments performed in triplicate. Significance of difference: in (A): * p<0.05, ** p<0.01 vs WT PM; ° p<0.05, °° p<0.01 vs SCR-siRNA PM (T0); ## p<0.01 vs WT DM 24h; §§ p<0.01 vs SCR-siRNA DM 24h. In (I,V-X): ** p<0.01 vs WT; °° p<0.01 vs SCR-siRNA (One-way ANOVA with post-hoc Tukey HSD).
Figure 8.
Evaluation of MMP-9 silencing or inhibition on C2C12 myoblast MyoD, Notch-1 and Ki67 expression and morphology. The cells were transfected in the proliferation medium (PM) with either scrambled siRNA (SCR-siRNA) or MMP-9-siRNA. After transfection - PM (T0) - the cells were induced to differentiate by shifting in the differentiation medium (DM) for 24 h. In some experiments the cells were cultured in DM in the presence of the MMP-9 inhibitor SB-3CT (10 µM). WT: wild type not-transfected or untreated cells. (A) Western Blotting analysis. Representative blot and bar charts showing the densitometric analysis of the bands normalized to α-tubulin. (B-E) Representative differential interference contrast (DIC) images of fixed C2C12 cells in the indicated experimental conditions. Scale bar: 12.5 µm. (F-H,J-U) Representative confocal fluorescence images of the cells immunostained with antibodies against the indicated markers (green). Scale bar: 25 µm. (I,X) Bar charts showing the densitometric analysis of the MMP-9 and Notch-1 fluorescent signal intensity (F.I.) performed on digitized images in 10 regions of interest (ROI) of 100 μm2 for each confocal stack (10). a.u.: arbitrary units. (V,W) Quantitative analyses of Ki67 and MyoD positive nuclei expressed as the percentage of the total nuclei, respectively. Data shown are mean ± SD and represent the results of at least three independent experiments performed in triplicate. Significance of difference: in (A): * p<0.05, ** p<0.01 vs WT PM; ° p<0.05, °° p<0.01 vs SCR-siRNA PM (T0); ## p<0.01 vs WT DM 24h; §§ p<0.01 vs SCR-siRNA DM 24h. In (I,V-X): ** p<0.01 vs WT; °° p<0.01 vs SCR-siRNA (One-way ANOVA with post-hoc Tukey HSD).

Figure 9.
Electrophysiological analysis. The cells were transfected in the proliferation medium (PM) with either scrambled siRNA (SCR-siRNA) or HIF-1α-siRNA. After transfection -PM (T0)- the cells were induced to differentiate by shifting in the differentiation medium (DM) for 24 h. WT: wild type, not-transfected or untreated cells. In other experiments the cells were cultured in PM or DM in the presence of the MMP-9 inhibitor SB-3CT (10 µM) for 24 h. (A) Pulse protocol of stimulation in voltage clamp (top) and typical tracings of passive membrane currents (bottom) obtained in response to its application. (B) Cell capacitance (Cm, in pF) as an index of cell surface. (C) Specific membrane conductance, Gm/Cm (in nS/pF). Data are mean ± SD. * p<0.05 vs WT PM, # p<0.05 vs WT DM 24h (One way ANOVA followed by Bonferroni's post hoc test).
Figure 9.
Electrophysiological analysis. The cells were transfected in the proliferation medium (PM) with either scrambled siRNA (SCR-siRNA) or HIF-1α-siRNA. After transfection -PM (T0)- the cells were induced to differentiate by shifting in the differentiation medium (DM) for 24 h. WT: wild type, not-transfected or untreated cells. In other experiments the cells were cultured in PM or DM in the presence of the MMP-9 inhibitor SB-3CT (10 µM) for 24 h. (A) Pulse protocol of stimulation in voltage clamp (top) and typical tracings of passive membrane currents (bottom) obtained in response to its application. (B) Cell capacitance (Cm, in pF) as an index of cell surface. (C) Specific membrane conductance, Gm/Cm (in nS/pF). Data are mean ± SD. * p<0.05 vs WT PM, # p<0.05 vs WT DM 24h (One way ANOVA followed by Bonferroni's post hoc test).
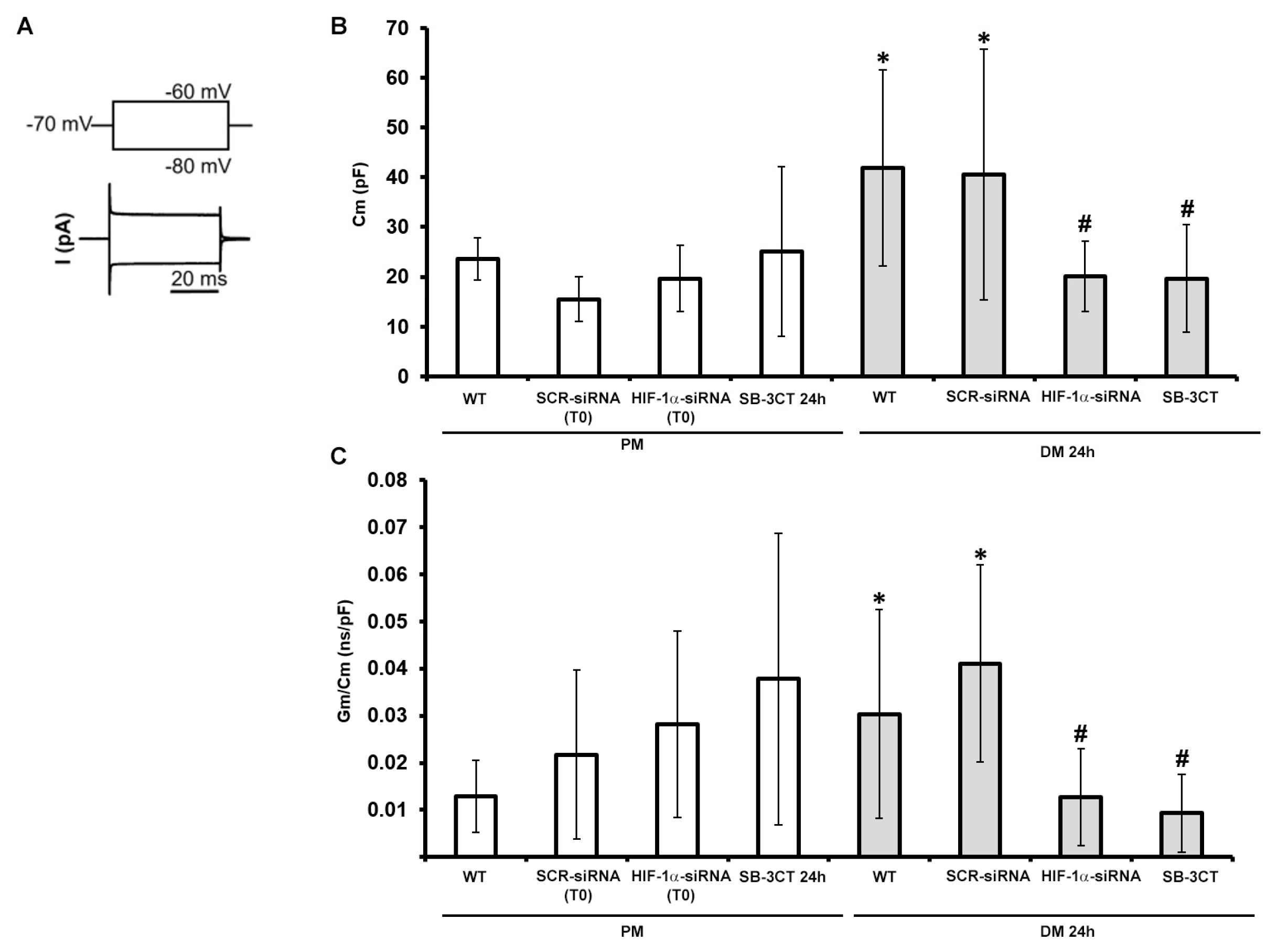
Figure 10.
Ca2+ current evaluation in C2C12 cells. Representative time course of Ca2+ currents normalized for cell capacitance (in pA/pF) from (A) a wild type (WT) C2C12 cell cultured in proliferation medium PM or (E) differentiation medium DM for 24 h; (B) from a scrambled siRNA (SCR-siRNA) transfected cell cultured in PM (T0) or (F) in DM for 24 h; (C) from a HIF-1α-siRNA transfected cell cultured in PM (T0) or (G) in DM for 24 h; (D) from a cell treated with the MMP-9 inhibitor SB-3CT (10 µM) for 24 h in PM or (H) in DM . (I) Pulse protocol of stimulation used to evoke ion currents. (J) Overall I-V relation related to the four conditions for all the experiments done in PM (WT PM, n=7, open circles ; SCR-siRNA PM (T0) n=4, open squares; HIF-1α-siRNA PM (T0) n=4, open triangles; SB-3CT PM 24h n=6, open diamonds) and (K) in DM (WT DM 24h n=6, gray circles; SCR-siRNA DM 24h n=2, gray squares; HIF-1α-siRNA DM 24h n=3, gray triangles; SB-3CT DM 24h n=7, gray diamonds). Data are mean ± SD. (p>0.05 two-way ANOVA).
Figure 10.
Ca2+ current evaluation in C2C12 cells. Representative time course of Ca2+ currents normalized for cell capacitance (in pA/pF) from (A) a wild type (WT) C2C12 cell cultured in proliferation medium PM or (E) differentiation medium DM for 24 h; (B) from a scrambled siRNA (SCR-siRNA) transfected cell cultured in PM (T0) or (F) in DM for 24 h; (C) from a HIF-1α-siRNA transfected cell cultured in PM (T0) or (G) in DM for 24 h; (D) from a cell treated with the MMP-9 inhibitor SB-3CT (10 µM) for 24 h in PM or (H) in DM . (I) Pulse protocol of stimulation used to evoke ion currents. (J) Overall I-V relation related to the four conditions for all the experiments done in PM (WT PM, n=7, open circles ; SCR-siRNA PM (T0) n=4, open squares; HIF-1α-siRNA PM (T0) n=4, open triangles; SB-3CT PM 24h n=6, open diamonds) and (K) in DM (WT DM 24h n=6, gray circles; SCR-siRNA DM 24h n=2, gray squares; HIF-1α-siRNA DM 24h n=3, gray triangles; SB-3CT DM 24h n=7, gray diamonds). Data are mean ± SD. (p>0.05 two-way ANOVA).
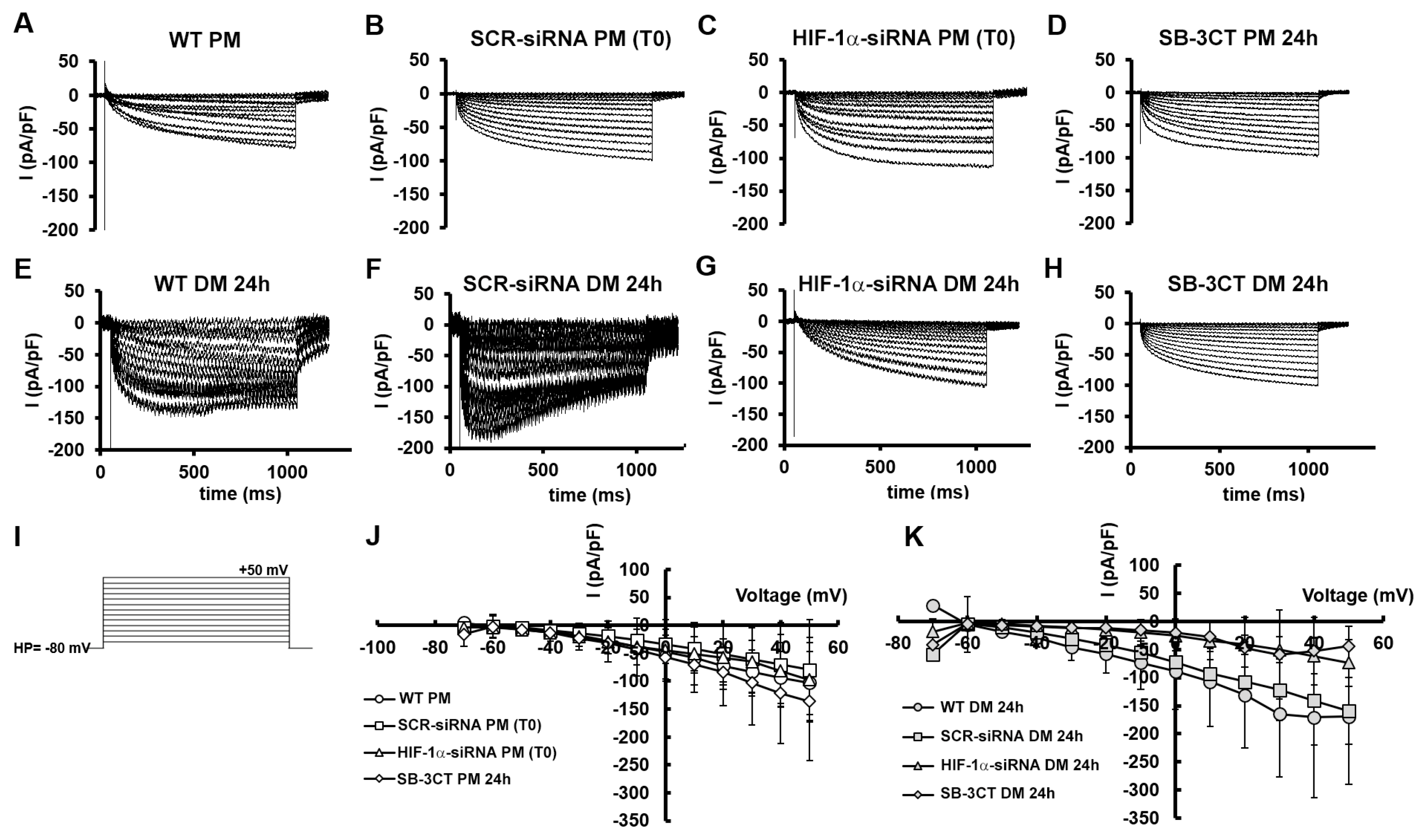

Table 1.
Specifications of primary antibodies used for immunofluorescence (IF) and Western Blotting (WB) analyses.
Table 1.
Specifications of primary antibodies used for immunofluorescence (IF) and Western Blotting (WB) analyses.
| Primary Antibody name | Company | Code | Dilution |
|---|---|---|---|
| anti-Ki67 rabbit polyclonal |
Abcam, Cambridge, UK | ab15580 | 1:100 (IF) |
| anti-Notch-1 rabbit monoclonal |
Abcam | ab52627 | 1:100 (IF) 1:1000 (WB) |
| anti-MyoD (M-318) rabbit polyclonal |
Santa Cruz Biotechnology |
sc-760 | 1:50 (IF) 1:500 (WB) |
| anti-myogenin (F5D) mouse monoclonal |
Santa Cruz Biotechnology |
sc-12732 | 1:50 (IF) |
| anti-α-sarcomeric actin mouse monoclonal |
DakoCytomation, Carpinteria, CA, USA |
M 0874 | 1:100 (IF) |
| anti- peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1α mouse monoclonal |
Santa Cruz Biotechnology |
sc-518025 | 1:100 (IF) |
| anti-HIF-1α rabbit polyclonal |
Santa Cruz Biotechnology |
sc-10790 | 1:100 (IF) 1:1000 (WB) |
| anti-MMP-9 rabbit polyclonal |
Bioss Antibodies, Woburn, MA, USA |
bs-0397R | 1:100 (IF) 1:1000 (WB) |
| anti-Pax7 mouse monoclonal |
Santa Cruz Biotechnology |
sc-81648 | 1:100 (IF) |
| anti-succinate dehydrogenase complex iron sulfur subunit-B (SDH-B) rabbit polyclonal |
Sigma-Aldrich | SAB2102103 | 1:1000 (WB) |
| anti-lactate dehydrogenase (LDH)-A (E-9) mouse monoclonal |
Santa Cruz Biotechnology |
sc-137243 | 1:1000 (WB) |
| anti-α-tubulin rabbit polyclonal |
GeneTex, Prodotti Gianni, Milano, Italy | GTX112141 | 1:1000 (WB) |
Table 2.
C2C12 cells passive membrane properties. The cells were treated and cultured as indicated in Figure 9. At least three independent C2C12 cells culture preparation were analyzed. n: number of the cells. Data are mean ± SD. * p<0.05 vs WT PM; # p<0.05 vs WT DM 24h (One way ANOVA followed by Bonferroni's post hoc test).
Table 2.
C2C12 cells passive membrane properties. The cells were treated and cultured as indicated in Figure 9. At least three independent C2C12 cells culture preparation were analyzed. n: number of the cells. Data are mean ± SD. * p<0.05 vs WT PM; # p<0.05 vs WT DM 24h (One way ANOVA followed by Bonferroni's post hoc test).
| Condition | Cm (pF) | Gm/Cm (nS/pF) |
|---|---|---|
| WT PM | 23.56±4.20 n=5 |
1.29×10-2±0.77×10-2 n=10 |
| SCR-siRNA PM (T0) |
15.51±4.43 n=3 |
2.18×10-2±1.79×10-2 n=4 |
| HIF-1α-siRNA PM (T0) |
19.65±6.68 n=4 |
2.82×10-2±1.97×10-2 n=4 |
| SB-3CT PM 24h | 25.08±17.06 n=6 |
3.78×10-2±3.10×10-2 n=5 |
| WT DM 24h | 41.87±19.62* n=10 |
3.04×10-2±2.21×10-2* n=10 |
| SCR-siRNA DM 24h |
40.58±25.22* n=7 |
4.11×10-2±2,09×10-2* n=6 |
| HIF-1α-siRNA DM 24h |
20.08±7.02# n=5 |
1.27×10-2±1.03×10-2# n=11 |
| SB-3CT DM 24h |
19.68±10.81# n=5 |
0.93×10-2±0.82×10-2# n=4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.


HIF-1α/MMP-9 Axis Is Required for the Early Phases of Skeletal Myoblast Differentiation under Normoxia Condition In Vitro
Flaminia Chellini
et al.
,
2023
The DUX4-HIF1α Axis in Murine and Human Muscle Cells: A Link More Complex than Expected
Thuy-Hang Nguyen
et al.
,
2024



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated