
Preprint
Article
Selenium-Containing (Hetero)Aryl Hybrids as Potential Antileishmanial Drug Candidates: In vitro Screening Against L. amazonensis
Altmetrics
Downloads
105
Views
37
Comments
0
A peer-reviewed article of this preprint also exists.
supplementary.pdf (277.34KB )






This version is not peer-reviewed
Submitted:
20 December 2023
Posted:
21 December 2023
You are already at the latest version
Alerts
Abstract
Leishmaniasis remains a significant global health concern, with current treatments relying on outdated drugs associated with high toxicity, lengthy administration, elevated costs, and drug resistance. Consequently, the urgent need for safer and more effective therapeutic options in leishmaniasis treatment persists. Previous research has highlighted selenium compounds as promising candidates for innovative leishmaniasis therapy. In light of this, a library of 10 selenium-containing diverse compounds was designed and evaluated in this study. The compounds were subjected to screening against L. amazonensis promastigotes, and their cytotoxicity was assessed in NIH/3T3 cells. Among the tested compounds, MRK-106 and MRK-108 displayed the highest potency against L. amazonensis promastigotes while demonstrating reduced cytotoxicity. Notably, MRK-106 and MRK-108 exhibited IC50 values of 3.97 µM, 4.23 µM, respectively, and most of the tested compounds showed low cytotoxicity in NIH/3T3 cells (CC50 > 200 µM). Although these results are promising, additional investigations focusing on intracellular amastigotes are required to determine the antileishmanial activity of the compounds. In conclusion, the identified selenium-containing compounds hold considerable potential as antileishmanial drug candidates and warrant further exploration in subsequent studies. These findings represent a significant step towards the development of safer and more effective therapies for leishmaniasis, addressing the pressing need for novel and improved treatments.
Keywords:
Subject: Medicine and Pharmacology - Other
1. Introduction
Leishmaniases encompass a diverse spectrum of diseases, each presenting a wide array of clinical manifestations. These clinical conditions are attributed to various species of the kinetoplastid parasite Leishmania, affecting both humans and other mammals dwelling in tropical and subtropical regions worldwide [1,2]. According to the World Health Organization (WHO), today, more than 1 billion people live in areas endemic for leishmaniasis and are at risk of infection. An estimated 30 000 new cases of VL and more than 1 million new cases of CL occur annually, with an ongoing burden of 12 million people harboring active infections [3]. The transmission of this pathology is facilitated by blood-feeding female sandflies, with the parasites undergoing a complex life cycle involving two distinct forms: extracellular flagellated promastigotes within the vector and intracellular non-flagellated amastigotes residing within mononuclear phagocytes in the mammalian host [4].
Historically, pentavalent antimony stood as the primary treatment for leishmaniasis. However, it comes burdened with concerns of cardiotoxicity, cirrhosis, pancreatic toxicity, and the risk of resistance development [5]. Consequently, amphotericin B (including lipid formulations) emerged as a secondary option. The repertoire of drugs repurposed for leishmaniasis treatment includes amphotericin B, miltefosine, paromomycin, and pentamidine [6]. Miltefosine has also found its place in treating both VL and CL, offering the benefits of oral administration, high efficacy, and a short treatment course. Nonetheless, its usage is hampered by teratogenicity and the potential for drug resistance [6,7,8]. Hence, there is an immediate need to discover new therapeutic approaches and drug compounds to combat these life-threatening diseases.
Selenium compounds are gaining remarkable prominence in medicinal chemistry, representing a burgeoning frontier in the search for novel antiprotozoal agents [9,10]. The strategic integration of selenium (Se) atoms into organic frameworks offers a highly auspicious avenue for the creation of enhanced, disease-specific compounds. Organoselenium compounds are renowned for their diverse pharmacological properties [9,10,11,12,13,14,15,16,17,18,19,20]. Furthermore, the biocompatibility, minimal toxicity, and chemical versatility of selenium have spurred the development of a diverse range of Se-based pharmaceuticals. Research has demonstrated that the incorporation of selenium atoms into small molecules significantly amplifies their bioactivity [21]. In broad terms, organoselenium compounds exhibit medical applications that encompass cancer treatment, managing infections, inflammation, addressing Alzheimer’s disease and depression, as well as providing antioxidant benefits [21]. Intriguingly, selenium has also exhibited favorable effects in combatting parasitic diseases, including but not limited to malaria, African trypanosomiasis, Chagas Disease and intestinal parasites [22]. This highlights the pivotal role of selenium compounds in the pursuit of effective treatments against this parasitic disease [22,23,24]. Various Se-containing compounds have shown antimicrobial properties. Additionally, studies indicate that Se supplementation can reduce parasite burden and ameliorate symptoms associated with Leishmania spp. and other trypanosomiasis [22]. Moreover, recent findings have highlighted the in vitro leishmanicidal potential of newly synthesized compounds featuring selenium within their structures.
Similarly, heterocyclic compounds hold a pivotal role in organic chemistry, primarily due to their ubiquitous presence in pharmaceuticals, natural substances, and various chemicals integral to our daily lives [25,26,27,28,29,30]. These compounds are characterized by the presence of one or more heteroatoms within cyclic structures, with or without aromatic properties. Oxygen, nitrogen, phosphorus, and sulfur rank among the most frequently incorporated heteroatoms in a majority of heterocyclic compounds [32]. Around, 80% of major commercially available synthetic drugs contain at least one heterocyclic scaffold, with a broad spectrum of pharmacological potential, encompassing applications such as antitumor, anti-inflammatory, and especially important for several active compounds against microorganisms [31]. In the past few decades, the quest for safer and more efficacious drugs to combat leishmaniasis has spurred a multitude of research initiatives. Researchers worldwide have dedicated extensive efforts to synthesize a wide array of antileishmanial agents, distinguished by their incorporation of diverse heterocyclic moieties [33]. Among these moieties, thiazoles, pyrazoles, pyrimidines, chromanones, and imidazoles stand out, each offering a unique chemical scaffold that has garnered attention in the development of potential antileishmanial therapeutics. [31,32,33]. Considering the biological importance of heteroarenes and the wide spectrum of therapeutic properties of organoselenium compounds, molecular hybridization of these structures demonstrates promising biological properties.
Thus, in line with our continuous interest in discovering and developing new sustainable, efficient methodologies for biologically relevant organoselenides and their biological evaluation [17,19,34,35,36,37,38], this article aims to thoroughly examine the synthetic approaches used to create novel chemical compounds for combating leishmaniasis. Herein we report the in vitro investigation of antileishmanial potential of selenium substituted (hetero)aryl hybrids (indole, coumarin, chromone, oxadiazole, imidazo[1,2-a]pyridine, Imidazo[2,1-b]thiazole and oxazole, among others) seeking to assess their efficacy and potential for treating this parasitic disease, with a focus on understanding the relationships between compound structures and their biological activity. Our goal is to contribute to the broader understanding of innovative drug development for addressing this widespread tropical disease.
2. Materials and Methods
2.1. Synthesis of selenium substituted (hetero)aryl hybrids:
For the current study, we selected several nitrogen and oxygen containing heterocycles. Some of privileged naturally occurring heterocycles have also been selected, for example, indole, coumarin and flavanone. Besides, oxadiazole, imidazo[1,2-a]pyridine, Imidazo[2,1-b]thiazole and oxazole were also been selected. These heterocyclic compounds have been utilized to access structurally diverse selenium substituted (hetero)aryl hybrids MRK101-108, 111, 113 (Figure 1) using our previously established sustainable routes. [34,35,36,37,38,39,40].
2.2. Mice and parasites
Female BALB/c mice, aged 6-8 weeks with an average weight of 30 g, were obtained from the Central Animal Facility of the Federal University of Mato Grosso do Sul (UFMS, Campo Grande-MS, Brazil). Animals were housed in individually ventilated cages (IVCs) within a ventilated rack system (Alesco) under specific pathogen-free (SPF) conditions. The environment maintained a temperature of 25°C ± 1°C, a 12-hour light/dark cycle, and ad libitum access to Nuvital food and water. All procedures were approved by the Ethics Committee on Animal Experiments (CEUA) of UFMS (protocol number 1.041/2019) and adhered to relevant guidelines for animal welfare and research.
Leishmania (Leishmania) amazonensis (IFLA/BR/1967/PH8 strain) were maintained as promastigotes at 26°C in Schneider’s medium (Sigma-Aldrich) supplemented with 20% heat-inactivated fetal bovine serum (FBS, Sigma-Aldrich), 10,000 U/mL penicillin, and 10 mg/mL streptomycin (Sigma-Aldrich) for a maximum of 20 serial passages. Parasites in the exponential growth phase were used for the in vitro antipromastigote assay, while those in the stationary phase were used for the antiamastigote assay. Intracellular amastigote forms were obtained by infecting murine peritoneal macrophages with promastigotes.
2.3. Anti-promastigotes assays:
Selenium-compounds MRK-101 to 113 were evaluated for their antipromastigote activity against Leishmania (Leishmania) amazonensis in five replicates. Serial dilutions ranging from 100, 50, 25 and 12.5 µM were prepared in supplemented Schneider’s Insect Medium (Sigma-Aldrich). The microplates were then seeded with L. amazonensis promastigotes (1×105 parasites/well) and incubated at 26°C for 48 hours in a Biochemical Oxygen Demand (BOD) incubator (Cienlab, Brazil). Cell viability was assessed using the resazurin (Sigma-Aldrich) assay. A solution of resazurin (Sigma-Aldrich, USA) at 0.2 mg.mL-1 was added to each well, and after 4 h of incubation at 26oC, the absorbances at 570 nm and 600 nm were acquired and the viability calculation for all wells was performed based on the formula and instructions provided by the AlamarBlue® manufacturer’s website [41]. Cell viability was evaluated from optical density and compared to untreated cells. Cells incubated with amphotericin B (1.25, 2.5, 5 and 10 µM) served as reference antipromastigote drug, while DMSO in Schneider’s Insect Medium (Sigma-Aldrich) was used as the negative control. The half-maximal inhibitory concentration (IC50) values for each compound were calculated using a nonlinear dose-response regression curve generated Prism 5 (GraphPad software, Boston, MA). To compare variances, ANOVA with Tukey’s posttest was used, supplemented by the parametric t test, with a 95% confidence interval.
2.4. Peritoneal macrophages
Following animal euthanasia, murine peritoneal macrophages were harvested. Ten milliliters of cold RPMI 1640 medium (Sigma-Aldrich) supplemented with 10,000 U/mL penicillin and 10 mg/mL streptomycin (Sigma-Aldrich) was injected into the peritoneal cavity. The abdominal region was gently massaged to facilitate macrophage release. The peritoneal fluid was then aspirated and transferred to an ice-cold erlenmeyer flask to prevent cell adherence. Cell quantification was performed using a Neubauer’s chamber after trypan blue staining (Sigma-Aldrich) for viability assessment.
2.5. Treatment of infected macrophages
Murine peritoneal macrophages (1x106 cells/well) were seeded in three replicates onto 24-well plates containing round glass coverslips (13 mm) pre-coated with 10% FBS in RPMI 1640 medium (Sigma-Aldrich). Plates were incubated at 37°C with 5% CO2 for 1 hour to allow adhesion, confirmed by microscopy. Following two washes with PBS (Sigma-Aldrich), adherent macrophages were infected with stationary-phase L. amazonensis promastigotes (4x106 parasites/mL) and incubated at 35°C/5% CO2. After 4 hours, free parasites were removed by two PBS washes, and infected cells were treated for 24 hours with the compounds at concentrations 6.25, 12.5, 25 and 50 μM. Amphotericin B (Sigma-Aldrich; 1.25, 2.5, 5 and 10 μM) served as reference drug, and untreated cells were used as a negative control. Supernatants were removed after treatment, and cells were washed twice with PBS, fixed with Bouin’s solution (Sigma-Aldrich), and stained with Giemsa (Sigma-Aldrich) diluted 1:10 in distilled water. Coverslips were dehydrated through an acetone/xylene gradient (100%, 70%, 50%, 30%, 100% each with Sigma-Aldrich) and mounted for microscopic visualization. The total number of intracellular amastigotes was counted in 200 cells per coverslip in three replicates using an optical microscope. The half-maximal inhibitory concentration (IC50) values were calculated for each compound using a nonlinear regression curve in GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA, USA).
2.6. Cytotoxicity assays:
Murine peritoneal macrophages (1x106 cells/well), NIH/3T3 cells (ATCC CRL-1658, mouse fibroblasts lineage) and J774A.1 (ATCC, mouse macrophages lineage) were cultured in 96-well plates (1x105 cells/well) in RPMI 1640 medium (Sigma-Aldrich, USA) supplemented with 10% fetal bovine serum (Cultilab, Brazil), 10 IU.mL-1 of penicillin, and 100 µg.mL-1 of streptomycin (Sigma-Aldrich, USA) and adhered overnight at 37oC and 5% CO2. The medium was replaced by fresh RPMI medium with different concentrations of the compounds ranging from 200 to 3.12 µM and incubated for 48 h. Cells treated with amphotericin B (50.0–0.78 µM) served as the antileishmanial drug reference. Dimethyl sulfoxide (DMSO, Sigma-Aldrich) at the concentration required to solubilize the highest test sample concentration was used as control and did not affect cell viability control, as well as fibroblasts cultured with medium alone were used as a life/viability control. Cell viability was assessed using the colorimetric resazurin method [41]. Then, 5 µl of 0.2 mg.mL-1 resazurin solution (Sigma-Aldrich, USA) was added to each well, incubated for 4 h 37oC and 5% CO2. Absorbances were acquired at 570 and 600 nm and the viability calculation were performed as described in Section 2.3. The half-maximal cytotoxic concentration (CC50) for each compound was calculated from a sigmoidal regression of the dose-response curve generated using GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA, USA). In order to compare variances, ANOVA with Tukey´s posttest was used, supplemented by the parametric t test, with a 95% confidence interval.
3. Results
3.1. Anti-promastigotes and anti-amastigotes assays:
The antileishmanial activity of the synthesized selenium compounds were evaluated by culturing promastigote forms of L. amazonensis in the presence of different concentrations of each molecule. Control wells containing only the culture medium (C) and amphotericin B (ANFB), a reference drug for antileishmanial activity, were also prepared. After 48 h of incubation, the percentage of promastigote form’s viability was accessed by the resazurin colorimetric method and calculated comparing to control wells. The antileishmanial activity of the selenium compounds were also assessed in intracellular amastigote forms inside infected murine peritoneal macrophages and the representative results obtained for five of the ten compounds tested are illustrated in Figure 2.
The half-maximal inhibitory concentration (IC50) of each compound was obtained by nonlinear regression and the results are shown in Table 1. The IC50 of tested compounds ranged from 3.96 to 40.98 µM for promastigote forms, and from 15.93 to >50 µM for amastigote forms. Amphotericin B, a reference drug for its anti-leishmanial activity, was also evaluated under the used experimental conditions, obtaining an IC50 of 9.40 µM for promastigotes and 0,97 µM for intracellular amastigote forms.
The antileishmanial activity of selenium-compounds was evaluated against promastigote and amastigotes forms of L. amazonensis after 48 and 24 hours, respectively. The cytotoxicity was performed in BALB/c peritoneal macrophages. Selectivity index (SI) was calculated as CC50/IC50 values for promastigote and amastigote forms, respectively. Abbreviations: CC50= half-maximal cytotoxic concentration; IC50 = half-maximal inhibitory concentration; SI = selectivity index. aSI, CC50 on murine peritoneal macrophages / IC50 on promastigotes. bSI, IC50 on murine peritoneal macrophages / IC50 on amastigotes. ANFB = amphotericin B used as reference drug for L. amazonensis. Ind = means indetermined results.
The highest activity against promastigote forms of L. amazonensis was observed for compound MRK-106, which contains a 4,5-dihydrooxazole ring in its structure and showed an IC50 of 3.96 µM. Compound MRK-108, a selenocyanate linked to a coumarin ring, had an IC50 of 4.23 µM, the second highest potency observed against the parasite. Following the decreasing order of anti-leishmania activity observed, the following compounds showed similar activity, between 10 and 20 µM: MRK-105, a phenol selenocyanate, showed an IC50 of 12.17 µM; MRK-102, a beta-naphthol, showed an IC50 of 15.15 µM; compound MRK-103, which has a 1,3,4-oxadiazole ring, with an IC50 of 15.48 µM; MRK-104, which has an indolic nucleus, showed an IC50 of 16.17 µM, and compound MRK-111, an imidazothiazole, with an IC50 of 17.55 µM. The least active compounds had IC50s above 20 µM: MRK-107, an imidazopyridine, with an IC50 of 27.37 µM; MRK-101, a chromone, with an IC50 of 30.46 µM and MRK-113, also an imidazopyridine, with an IC50 of 40.98 µM.
It is interesting to note that the activity of the compounds against promastigote forms did not necessarily reflect the results of the anti-amastigote intracellular activity experiments: the compounds MRK-106 and MRK-108 were not active in macrophages infected with L. amazonensis, with IC50 values estimated at >50 µM, the highest concentration tested. On the other hand, the compounds MRK-107 and MRK-113, which were poorly active against promastigote forms, showed the best results against intracellular amastigote forms: 18.31 and 15.93 µM, respectively. As most of tested compounds did not showed activity against intracellular amastigotes, their SI were considered indetermined (as indicated as ind in Table 1).
3.2. Cytotoxicity and selectivity index:
The selectivity index (SI) is defined as the ratio of the CC50 obtained for host cells to the IC50 obtained against the parasite cells and helps identify the compounds that exhibit a high degree of specificity in targeting the pathogen of interest while sparing host cells [42,43]. To determine the selectivity index of the selenium compounds, their cytotoxicity on murine peritoneal macrophages, NIH/3T3 murine fibroblast cell line and J774A.1 murine macrophages cell line was also evaluated, at concentrations ranging from 200 to 3.12 µM. After 48 h, viability was assessed using the resazurin colorimetric methodology, where cells incubated only with culture medium were considered 100% viable. The representative results obtained for five of the ten compounds tested are illustrated in Figure 2. The CC50 was obtained by non-linear regression and the results for CC50 for peritoneal macrophages are also shown in Table 1. CC50 and SI results for NIH/3T3 and J774A.1 cells for each compound are shown in Supplemental Material.
Figure 3.
Cytotoxicity of synthesized selenium compounds MRK-105, 106, 107, 108 and 113 against NIH/3T3 fibroblasts (A, B, C, D and E upper graphs), J774A.1 macrophages (F, G, H, I and J middle graphs) and murine peritoneal macrophages (K, L, M, N and O lower graphs). * and *** means p< 0,01 and p< 0,001, respectively (ANOVA and Tukey´s post test).
Figure 3.
Cytotoxicity of synthesized selenium compounds MRK-105, 106, 107, 108 and 113 against NIH/3T3 fibroblasts (A, B, C, D and E upper graphs), J774A.1 macrophages (F, G, H, I and J middle graphs) and murine peritoneal macrophages (K, L, M, N and O lower graphs). * and *** means p< 0,01 and p< 0,001, respectively (ANOVA and Tukey´s post test).

All the tested selenium compounds showed a CC50 above 200 µM in peritoneal macrophages. Some compounds showed higher cytotoxicity in other cell lines: compound MRK-105, whose CC50 was 87.85 µM in NIH/3T3 cells and 60.16 µM in J774A.1 cells, and compound 104 with CC50 of 126.7 µM in J774A.1 cells. Thus, the most active compounds against L. amazonensis promastigotes were those compounds with the higher SI, compounds MRK-106 and MRK-108, with SI above 50.53 and 47.31, respectively. However, considering the cytotoxicity in peritoneal macrophages and the activity against intracellular amastigotes of L. amazonensis, the most active compounds were MRK-113 and MRK-107, with SI values of 12.55 and 10.92, respectively.
4. Discussion
Leishmaniasis, a neglected tropical disease, continues to pose as a significant challenge for drug discovery and development. Current antileishmanial drugs have issues such as high toxicity, resistance development, and the necessity for hospitalization, rendering them poor adhesion [3,4]. While there have been notable strides made through combination therapy approaches, which have reduced treatment duration and cost, a critical gap persists — the urgent need for new active drugs. In the search for novel drugs, emerging evidence highlights a link between selenium and parasites, notably trypanosomatids. Certain parasites have been found to both express selenoproteins and metabolize selenium. This underscores the potential significance of selenium as a promising element for the development of new agents against leishmaniasis. Here, we demonstrated that using sustainable routes, synthesized (hetero)aryl hybrids selenium-compounds displayed anti-promastigote activity in vitro and a promising selectivity index, with MRK-106, MRK-107, MRK-108 and MRK-113 being the most potent and selective for antileishmanial activity.
MRK-106 contains a 4,5-dihydrooxazole ring (oxazoline), with substituents containing benzene rings in positions 2 and 5, showed an IC50 of 3.96 µM for promastigotes and a selectivity index of 50.53. Secondary metabolites featuring oxazole, oxazoline, and isoxazoline ring structures exhibit a broad distribution across marine and terrestrial organisms [44,45]. Among various heterocyclic compounds, isoxazoles and their analogs hold great significance due to their wide-ranging biological activities. This makes them pivotal structures in medicinal chemistry. Isoxazole derivatives with structural variations exhibit diverse medicinal properties, contributing significantly to the development of novel, highly effective, and less toxic bioactive drugs. These compounds are notable for their diverse and substantial biological activities, encompassing anti-tumor, antibacterial, anti-viral, anti-malarial, and immunosuppressive properties, including antileishmanial activity [44]. Moraski and colleagues undertook the synthesis of multiple compounds with oxazoline and oxazole motifs and tested for their inhibitory potential against Mycobacterium tuberculosis, showing promising results [45,46]. More recently, a series of compounds containing β-carboline-oxazoline were tested against promastigote and amastigote forms of L. amazonensis, and some of them were found to be active against the parasite [47]. Among them, compounds 8d and 8i were considered the most potent against promastigote forms, showing IC50 of 14,7 and 23 µM, and SI of 6.6 e 1,3, respectively. Our compound MRK-106 also have oxazoline ring in its structure, together with Se atom and two benzene rings, which resulted in a lower IC50 and higher SI, compared to the β-carboline-oxazoline molecules. Another compound containing an azol is MRK-103, which has a 1,3,4-oxadiazole ring. Despite several examples of the use of different azol moieties in active antileishmanial compounds [44], MRK-105 showed an IC50 of 15.48 µM and SI of 12.92, more than three times less selective than MRK-108. It is also interesting to note that, although a higher IC50 was obtained than for amphotericin B under the experimental conditions used, the selectivity index for MRK-106 (50.53) is more than five times higher than that obtained for this drug (9.40) for promastigote forms. Amphotericin B is an important second-choice drug in the treatment of leishmaniasis, but it is also known for its toxic side effects and low therapeutic index [3,4]. Thus, the SI obtained for MRK-106 is also important data for future studies of anti-leishmanial activity.
Compound MRK-108, a selenocyanate linked to a coumarin ring, showed an IC50 of 4.23 µM and an SI of 47.31. The potential for functionalization and distinctive attributes renders coumarin a privileged scaffold in the field of medicinal chemistry [48]. While coumarins are predominantly found as secondary metabolites in plants, bacteria, and fungi, numerous synthetic methods have been documented for their production. This bicyclic heterocycle, composed of a benzene ring fused with a pyrone ring, exhibits the capability to engage with diverse biological targets [48]. The pyrone ring facilitates hydrogen bonding with multiple amino acid residues, while the aromatic segment can establish hydrophobic interactions. Consequently, this versatility results in a wide array of biological properties, encompassing antioxidant, anticoagulant, anticancer, antiviral, antitrypanosomal, anticholinesterase, and antileishmanial activities. Coumarin derivatives were identified as promising structures in the search for new anti-leishmania agents in a recent review [49]. Also, the authors pointed out that the presence of electron withdrawing groups increase the antileishmanial effect. Besides, selenocyanates displayed potency against Leishmania infantum promastigotes [24]. In case of compound MRK-108, selenium atom is linked to nitrile (strongly electron-withdrawing group) [50,51] and on other side to methyl-2H-pyran-2-one of coumarin, and this may contribute to the activity observed for MRK-108. The authors also highlight the work of Huang and colleagues with quinolines derivatives containing selenium as promising antileishmanial candidates [49,51]. The SI obtained for MRK-108 (47.31) was also almost three times higher than that of amphotericin B, which could be a promising characteristic for the therapeutic use of this molecule, or a series of derivative compounds for further structure activity relationship studies.
MRK-107 and MRK-113 showed low activity against L. amazonensis promastigotes (27,37 and 40,98 µM, respectively), and it is interesting to observe that both are imidazopyridine compounds, with a phenyl-selenyl substituent in carbon 3. But the presence of a methoxy group for MRK-107 enhanced its activity almost 1,5x compared to MRK-113, without methoxy, an electron-donating group. This should contribute to the antileishmanial activity and, like other compounds studied here, further structure-related activity should be carried out to address the ligand and/or the essential radicals responsible for the observed activity. Despite the results obtained for promastigote cells, both MRK-107 and MRK-113 were the most active compounds against intracellular amastigotes, with IC50 of 18.31 and 15.93 µM and SI 12.55 and 10.92, respectively. The imidazopyridine scaffold has gained significant importance for designing synthetic analogs targeting a range of therapeutic disorders, including cancer, diabetes, infections, inflammation, and CNS conditions. This heterocyclic system serves as a crucial pharmacophore motif, expanding medicinal chemistry tools. Additionally, imidazopyridines find use in combating helminthic, coccidial, and fungal infections, illustrating their multifaceted role in drug development [53]. Recently, imidazopyridine derivatives are reported to have potential for anti-trypanosomiases drug discovery. Fersing et al. designed and synthesized novel 3-nitroimidazo[1,2-a]-pyridine derivatives. By introducing a heteroatom bridge between the aryl group and the imidazopyridine, they obtained the desired derivatives from 8-bromo-6-chloro-3-nitro-2-(phenylsulfonylmethyl)imidazo[1,2-a]pyridine. These compounds were tested in vitro against L. donovani and L. infantum strains, alongside reference drugs like pentamidine, fexinidazole, miltefosine, and amphotericin B [54]. Such structural features can serve as a basis for the design and synthesis of new series of imidazopyridines containing selenium.
Various chemical structures containing selenium have been investigated against Leishmania parasites, including diselenide, selenourea, methylseleno and selenocyanate components [22]. Both MRK-105 and MRK-108 contain selenocyanate radical, but their structures are quite different: MRK-105 is a phenol selenocyanate, whilst MRK-108 is selenocyanate linked to a coumarin, as described above. Also, MRK-8 showed higher activity against L. amazonensis promastigotes (4.23 µM) than MRK-105 (12.17 µM), despite its poor activity against intracellular amastigotes. Regarding selectivity index, MRK-105 was the only compound which cytotoxicity in NIH/3T3 fibroblasts was lower than 200 µM (87.85 µM), resulting in a low selectivity index: 7.22. Also, MRK-104 and MRK-105 showed higher cytotoxicity in J774A.1 cells: 126.7 and 60.16 µM, respectively. MRK-8, by its turn, exhibited a SI of 47.31. These results highlight the promising structure of selenocynate associated to coumarin ring in the search of novel antileishmanial drugs.
Based on the recommendations of the World Health Organization (WHO) Special Programme for Tropical Disease Research (TDR), some authors classify the compounds tested for L. donovani or L. infantum as active for an IC50 in amastigotes in macrophages of 1-2 µg/ml, moderately active for IC50 between 1.0 and 6.0 µM, and inactive for IC50 >6.0 µM, with a desirable SI>10 or even >20 [42,55,56]. This is an important step regarding antileishmanial potential, as intracellular amastigotes are the parasite forms found in mammalian host. However, testing new compounds against promastigote forms of Leishmania is still an important step in the search for their biological activity, as recently reviewed considering natural and synthetic compounds with antileishmanial activity [57,58]. So, further evaluation of these selenium compounds in other Leishmania species or even modifications on their structures should be carried out to outline their promising antileishmanial activity.
5. Conclusions
In conclusion, a series of selenium substituted (hetero)aryl hybrids were screened against L. amazonensis promastigotes and intracellular amastigotes. These hybrid compounds included selenium substituted indole, coumarin, chromone, oxadiazole, imidazo[1,2-a]pyridine, Imidazo[2,1-b]thiazole and oxazole, among others. The biological evaluation indicate that these hybrids are promising structures against this parasite. Furthermore, their cytotoxicity was also assessed in murine peritoneal macrophages, NIH/3T3 and J774A.1 cells, with good selectivity. Compounds containing oxazoline rings, coumarin derivatives and imidazopyridine rings showed the best parameters in the evaluation of antileishmanial activity and selectivity used. These compounds may serve as a basis for the synthesis of structural derivatives and thus develop more detailed structure activity studies. These data in this study represent an important step towards the search for new antileishmanial drugs to be further explored.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, E.B.P.; S.S.; J.R. and T.B.R.; methodology, M.H.F.; A.R.N.; M.S.A.B.; C.B.V.; A.L.S.; T.E.A.F. and C.C.P.A.; software, M.H.F.; A.R.N.; M.S.A.B.; C.B.V.; A.L.S. and C.C.P.A.; validation, M.H.F.; A.R.N.; M.S.A.B.; C.B.V.; A.L.S. and C.C.P.A.; formal analysis, M.H.F.; A.R.N.; M.S.A.B.; C.B.V.; A.L.S. and C.C.P.A.; investigation, A.L.B.; S.S.; J.R. and T.B.R.; resources, A.L.B.; S.S.; J.R. and T.B.R.; data curation, M.H.F.; A.R.N.; M.S.A.B.; C.B.V.; A.L.S.; T.E.A.F. and C.C.P.A.; writing—original draft preparation, S.S.; J.R. and T.B.R.; writing—review and editing, S.S.; J.R. and T.B.R.; visualization, J.R. and T.B.R.; supervision, S.S.; J.R. and T.B.R.; project administration, S.S.; J.R. and T.B.R.; funding acquisition, S.S.; J.R. and T.B.R.. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Finance Code 001) and Fundação de Apoio ao Desenvolvimento do Ensino, Ciência e Tecnologia do Estado de Mato Grosso do Sul (FUNDECT-MS). SS and JR are grateful to CNPq (315399/2020-1, 309975/2022-0, 422645/2021-4, 403210/2021-6, 404172/2023-7, and 405655/2023-1). S.S. would also like to acknowledge FAPEG (04/2023; EQU2023101000020).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We gratefully acknowledge CAPES (001), CNPq, UFMS, UFG, FUNDECT MS and INCT-Catálise/CNPq/FAPESC for the support offered in this research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Leishmaniasis. (n.d.). Available online: https://www.who.int/health-topics/leishmaniasis (accessed on 16 December 2023).
- Kmetiuk, L.B.; Tirado, T.C.; Bondo, L.M.; Biondo, A.W.; Figueirdo, F.B. Leishmania spp. in indigenous populations: A mini-review. Front. Public Health. 2022, 10, 1033803. [Google Scholar] [CrossRef] [PubMed]
- Altamura, F.; Rajesh, R.; Catta-Preta, M.C.; Moretti, N.S.; Cestari, I. The current drug discovery landscape for trypanosomiasis and leishmaniasis: Challenges and strategies to identify drug targets. Drug Dev. Res. 2020, 83, 225–252. [Google Scholar] [CrossRef] [PubMed]
- Torres-Guerrero, E.; Quintanilla-Cedillo, M.R.; Ruiz-Esmenjaud, J.; Arenas, R. Leishmaniasis: A review. F1000Research 2017, 6, 750. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, J.; Sundar, S. Current and emerging medications for the treatment of leishmaniasis. Expert Opin. Pharmacother. 2019, 20, 1251–1265. [Google Scholar] [CrossRef]
- Mazire, P.H.; Saha, B.; Roy, A. Immunotherapy for visceral leishmaniasis: A trapeze of balancing counteractive forces. Int. Immunopharmacol. 2022, 110, 108969. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Chakravarty, J. An update on pharmacotherapy for leishmaniasis. Expert Opin. Pharmacother. 2015, 16, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Verdan, M.; Taveira, I.; Lima, F.; Abreu, F.; Nico, D. Drugs and nanoformulations for the management of Leishmania infection: a patent and literature review (2015-2022). Expert Opin Ther Pat 2023, 33, 137–150. [Google Scholar] [CrossRef]
- Radomska, D.; Czarnomysy, R.; Radomski, D.; Bielawski, K. Selenium Compounds as Novel Potential Anticancer Agents. Int. J. Mol. Sci. 2021, 22, 1009. [Google Scholar] [CrossRef]
- Indira Priyadarsini, K.; G. Singh, B.; Kunwar, A. Current Developments on Synthesis, Redox Reactions and Biochemical Studies of Selenium Antioxidants. Curr. Chem. Biol. 2013, 7, 37–46. [Google Scholar] [CrossRef]
- Hoque, E.; Tran, P.; Jacobo, U.; Bergfeld, N.; Achary, S.; Shamshina, J.L.; Reid, T.W.; Abidi, N. Antimicrobial Coatings for Medical Textiles via Reactive Organo-Selenium Compounds. Molecules 2023, 28, 6381. [Google Scholar] [CrossRef]
- Hassan, A.A.; Kalinina, E.; Tatarskiy, V.; Shtil, A. The Thioredoxin System of Mammalian Cells and Its Modulators. Biomedicines 2022, 10, 1757. [Google Scholar] [CrossRef]
- Begines, P.; Martos, S.; Laguens, I.; Maya, I.; Padron, J.M.; Lopez, O.; Fernandes-Bolanos, J.G. Chemoselective Preparation of New Families of Phenolic-Organoselenium Hybrids—A Biological Assessment. Molecules 2022, 27, 1315. [Google Scholar] [CrossRef] [PubMed]
- da Costa, N.S.; Lima, L.S.; Oliveira, F.A.M.; Galiciolli, M.E.A.; Manzono, M.I.; Garlet, Q.I.; Irioda, A.C.; Oliveira, C.S. Antiproliferative Effect of Inorganic and Organic Selenium Compounds in Breast Cell Lines. Biomedicines 2023, 11, 1346. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xu, H. Incorporating Selenium into Heterocycles and Natural Products─From Chemical Properties to Pharmacological Activities. J. Med. Chem. 2022, 65, 4436–4456. [Google Scholar] [CrossRef] [PubMed]
- Sari, M.H.M.; Ferreira, L.M.; Prado, V.C.; Nogueira, C.W.; Cruz, L. Nano-based formulations as an approach for providing a novel identity for organoselenium compounds. Eur. J. Pharm. Biopharm. 2022, 178, 69. [Google Scholar] [CrossRef] [PubMed]
- Veloso, I.C.; Delanogare, E.; Machado, A.E.; Braga, S.P.; Rosa, G.K.; de Bem, A.F.; Rafique, J.; Saba, S.; da Trindade, R.N.; Galetto, F.Z.; Moreira, E.L.G. A selanylimidazopyridine (3-SePh-IP) reverses the prodepressant- and anxiogenic-like effects of a high-fat/high-fructose diet in mice. J. Pharm. Pharmacol. 2021, 73, 673. [Google Scholar] [CrossRef]
- Nie, Y.; Li, S.; Lu, Y.; Zhong, M.; Li, X.; Zhang, Y.; He, X. New Organoselenium (NSAIDs-Selenourea and Isoselenocyanate) Derivatives as Potential Antiproliferative Agents: Synthesis, Biological Evaluation and in Silico Calculations. Molecules 2022, 27, 4328. [Google Scholar] [CrossRef] [PubMed]
- Rafique, J.; Farias, G.; Saba, S.; Zapp, E.; Bellettini, I.C.; Momoli Salla, C.A.; Bechtold, I.H.; Scheide, M.R.; Santos Neto, J.S.; Souza Jr., D.M.; Braga, H.C.; Ribeiro, L.F.B.; Gastaldon, F.; Pich, C.T.; Frizon, T.E.A. Selenylated-oxadiazoles as promising DNA intercalators: Synthesis, electronic structure, DNA interaction and cleavage. Dyes Pigm. 2020, 180, 108519. [Google Scholar] [CrossRef] [PubMed]
- Begines, P.; Martos, S.; Lagunes, I.; Maya, I.; Padron, J.M.; Lopez, O.; Fernández-Bolaños, J.G. Chemoselective Preparation of New Families of Phenolic-Organoselenium Hybrids—A Biological Assessment. Molecules 2022, 27, 1315. [Google Scholar] [CrossRef]
- Chuai, H.; Zhang, S.-Q.; Bai, H.; Li, J.; Wang, Y.; Sun, J.; Wen, E.; Zhang, J.; Xin, M. Small molecule selenium-containing compounds: Recent development and therapeutic applications. Eur. J. Med. Chem. 2021, 223, 113621. [Google Scholar] [CrossRef]
- Alcolea, V.; Pérez-Silanes, S. Selenium as an interesting option for the treatment of Chagas disease: A review. Eur. J. Med. Chem. 2020, 206, 112673. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, S.; Fernández-Rubio, C.; Mansouri, R.; Ali-Hassanzadeh, M.; Ghani, E.; Karimazar, M.; Manzano-Román, R.; Nguewa, P. Selenium and protozoan parasitic infections: Selenocompounds and selenoproteins potential. Parasitol. Res. 2022, 121, 49. [Google Scholar] [CrossRef] [PubMed]
- Tieknik, E.R.T. Therapeutic potential of selenium and tellurium compounds: Opportunities yet unrealized. Dalton Trans. 2012, 41, 6390. [Google Scholar] [CrossRef] [PubMed]
- Aatif, M.; Raza, M.A.; Javed, K.; Nashre-ul-Islam, S.M.; Farhan, M.; Alam, M.W. Potential Nitrogen-Based Heterocyclic Compounds for Treating Infectious Diseases: A Literature Review. Antibiotics 2022, 11, 1750. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R. Introduction: Heterocycles. Chem. Rev. 2004, 104, 2125. [Google Scholar] [CrossRef]
- Franco, M.S.; Saba, S.; Rafique, J.; Braga, A.L. KIO4-mediated Selective Hydroxymethylation/Methylenation of Imidazo-Heteroarenes: A Greener Approach. Angew. Chem., Int. Ed. Engl. 2021, 60, 18454. [Google Scholar] [CrossRef]
- Ye, Z.; Adhikari, S.; Xia, Y.; Dai, M. Expedient syntheses of N-heterocycles via intermolecular amphoteric diamination of allenes. Nat. Commun. 2018, 9, 721. [Google Scholar] [CrossRef] [PubMed]
- Saba, S.; dos Santos, C.R.; Zavarise, B.R.; Naujorks, A.A.S.; Franco, M.S.; Schneider, A.R.; Scheide, M.R.; Affeldt, R.F.; Rafique, J.; Braga, A.L. Photoinduced, Direct C(sp2)−H Bond Azo Coupling of Imidazoheteroarenes and Imidazoanilines with Aryl Diazonium Salts Catalyzed by Eosin Y. Chem. Euro. J. 2020, 26, 4461. [Google Scholar] [CrossRef]
- Rizzo, C.; Amata, S.; Pibiri, I.; Pace, A.; Buscemi, S.; Piccionello, A.P. FDA-Approved Fluorinated Heterocyclic Drugs from 2016 to 2022. Int. J. Mol. Sci. 2023, 24, 7728. [Google Scholar] [CrossRef]
- Heravi, M.M.; Zadsirjan, V. Prescribed drugs containing nitrogen heterocycles: an overview. RSC Adv. 2020, 10, 44247. [Google Scholar] [CrossRef]
- Taylor, A.P.; Robinson, R.P.; Fobian, Y.M.; Blackemore, D.C.; Jones, L.H.; Fadeyi, O. Modern advances in heterocyclic chemistry in drug discovery. Org. Biomol. Chem. 2016, 14, 6611. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.N.; Jena, S.; Mukerjee, M.; Maiti, B.; Chanda, K. Green synthesis of biologically active heterocycles of medicinal importance: a review. Environ. Chem. Lett. 2021, 19, 3315. [Google Scholar]
- Scheide, M.R.; Peterle, M.M.; Saba, S.; Neto, J.S.S.; Lenze, G.F.; Cezar, R.D.; Felix, J.F.; Botteselle, G.V.; Schnedier, R.; Rafique, J.; Braga, A.L. Borophosphate glass as an active media for CuO nanoparticle growth: An efficient catalyst for selenylation of oxadiazoles and application in redox reactions. Sci. Rep. 2020, 10, 15233. [Google Scholar] [CrossRef]
- Neto, J.S.S.; Grangja, I.J.A.; Scheide, M.R.; France, M.S.; Moraes, C.A.O.; Beatriz, A.; de Lima, D.P.; Botteselle, G.V.; Frizon, T.E.A.; Saba, S.; Rafique, J.; Braga, A.L. Catalyst- and metal-free C(sp2)–H bond selenylation of (N-hetero)-arenes using diselenides and trichloroisocyanuric acid at room temperature. Sci. Rep. 2023, 13, 14251. [Google Scholar] [CrossRef]
- Doerner, C.V.; Neto, J.S.S.; Cabreira, C.R.; Saba, S.; Sandjo, L.P.; Rafique, J.; Braga, A.L.; de Assis, F.F. Synthesis of 3-selanyl-isoflavones from 2-hydroxyphenyl enaminones using trichloroisocyanuric acid (TCCA): a sustainable approach. New J. Chem. 2023, 47, 5598. [Google Scholar] [CrossRef]
- Peterle, M.M.; Scheide, M.R.; Silva, L.T.; Saba, S.; Rafique, J.; Braga, A.L. Copper-Catalyzed Three-Component Reaction of Oxadiazoles, Elemental Se/S and Aryl Iodides: Synthesis of Chalcogenyl (Se/S)-Oxadiazoles. ChemistrySelect 2019, 3, 13191. [Google Scholar] [CrossRef]
- Moraes, C.A.O.; Santos, R.B.C.; Cavalcante, M.F.O.; Guilhermi, J.S.; Ali, M.A.; Botteselle, G.V.; Frizon, T.E.A.; Shah, M.I.A.; Liao, L.M.; Beatriz, A.; Saba, S.; Rafique, J. Urea hydrogen peroxide (UHP) and Ethyl Lactate, an eco-friendly combo system in the direct C(sp2)-H bond selenylation of imidazo[2,1-b]thiazole and related structures. ACS Omega 2023. [Google Scholar] [CrossRef]
- Jacques, M.T.; de Souza, M.; Brabosa, F.A.R.; Canto, R.F.S.; Lopes, S.C.; Prediger, R.D.; Braga, A.L.; Aschner, M.; Farina, M. Novel Probucol Analogue, 4,4′-Diselanediylbis (2,6-Di-tert-Butylphenol), Prevents Oxidative Glutamate Neurotoxicity In Vitro and Confers Neuroprotection in a Rodent Model of Ischemic Stroke. ACS Chem. Neurosci. 2023, 14, 16–2857. [Google Scholar] [CrossRef]
- Corrêa, B. A., Síntese e avaliação do potencial antioxidante de cumarinas funcionalizadas com selênio, TCC Universidade Federal de Santa Catarina, Florianópolis-Brazil, 11/2015.
- Bio-Rad. Measuring cytotoxicity or proliferation - alamarBlue Assay Protocol. Bio-Rad [Internet]. Bio-Rad. Available online: https://www.bio-rad-antibodies.com/measuring-cytotoxicity-proliferation-spectrophotometry-fluorescence-alamarblue.html.
- Brioschi, M.B.C.; Coser, E.M.; Coelho, A.C.; Gadelha, F.R.; Miguel, D.C. Models for cytotoxicity screening of antileishmanial drugs: what has been done so far? Int. J. Antimicrob. Agents 2022, 60, 106612. [Google Scholar] [CrossRef]
- Álvarez-Bardón M, Pérez-Pertejo Y, Collazos A, Sepúlveda-Crespo D, Carballeira NM, Tekwani BL, et al. Screening Marine Natural Products for New Drug Leads against Trypanosomatids and Malaria. Mar. Drugs, 2020, 18, 187. [CrossRef]
- Kakkar, S.; Narasimhan, B. A comprehensive review on biological activities of oxazole derivatives. BMC Chem. 2019, 13, 16. [Google Scholar] [CrossRef]
- Moraski, G.C.; Chang, M.; Villegas-Estrada, A.; Franzblau, S.G.; Möllmann, U.; Miller, M.J. Structure–activity relationship of new anti-tuberculosis agents derived from oxazoline and oxazole benzyl esters. Euro. J. Med. Chem. 2020, 45, 1703. [Google Scholar] [CrossRef] [PubMed]
- Moraski, G.C.; Markley, L.D.; Chang, M.; Cho, S.; Franzblau, S.G.; Hwang, C.H.; Boshoff, H.; Miller, M.J. Generation and exploration of new classes of antitubercular agents: The optimization of oxazolines, oxazoles, thiazolines, thiazoles to imidazo[1,2-a]pyridines and isomeric 5,6-fused scaffolds. Bioorg. Med. Chem., 2012, 20, 2214. [Google Scholar] [CrossRef]
- Baréa, P.; de Paula, J.C.; Alonso, L.; de Oliveira, A.R.; da Costa, W.F.; Alonso, A.; Nakamura, C.V.; Sarragiotto, M.H. Synthesis, Antileishmanial Activity and Spin Labeling EPR Studies of Novel β-Carboline-Oxazoline and β-Carboline-Dihydrooxazine Derivatives. J. Braz. Chem. Soc. 2020, 31, 1170. [Google Scholar] [CrossRef]
- Gaudino, E.C.; Tagliapietra, S.; Martina, K.; Palmisano, G.; Cravotto, G. Recent advances and perspectives in the synthesis of bioactive coumarins. RSC Advances 2016, 6, 46394. [Google Scholar] [CrossRef]
- Gupta, O.; Pradhan, T.; Bhatia, R.; Monga, V. Recent advancements in anti-leishmanial research: Synthetic strategies and structural activity relationships. Euro. J. Med. Chem. 2021, 223, 113606. [Google Scholar] [CrossRef] [PubMed]
- Fourmigué, M.; Dhaka, A. Chalcogen bonding in crystalline diselenides and selenocyanates: From molecules of pharmaceutical interest to conducting materials. Coord. Chem. Rev. 2020, 403, 213084. [Google Scholar] [CrossRef]
- Jeannin, O.; Huynh, H.-T.; Riel, A.M.S.; Fourmigué, M. Chalcogen bonding interactions in organic selenocyanates: from cooperativity to chelation. New J. Chem. 2018, 42, 10502. [Google Scholar] [CrossRef]
- Huang, M.-F.N.; Luis, J.A.S.; da Silva, A.P.; Rocha, J.C.; Lima, T.K.S.; Scotti, M.T.; Scotti, L.; de Oliveira, R.F.; Souza, H.D.S.; de Athayde-Filho, P.F.; Barbosa-Filho, J.M. Synthesis, in silico Study and Antileishmanial Evaluation of New Selenides Derived from 7-Chloro-quinoline and N-Phenylacetamides. J. Braz. Chem. Soc. 2021, 32, 712–721. [Google Scholar] [CrossRef]
- Khatun, S.; Singh, A.; Bader, G.N.; Sofi, F.A. Imidazopyridine, a Promising Scaffold with Potential Medicinal Applications and Structural Activity Relationship (SAR): Recent Advances. J. Biomol. Struct. Dyn. 2021, 40, 14279. [Google Scholar] [CrossRef]
- Fersing, C.; Boudot, C.; Pedron, J.; Hutter, S.; Primas, N.; Castera-Ducros, C.; Bourgeade-Delmas, S.; Sournia-Saquet, A.; Moreau, A.; Cohen, A.; Stigliani, J.; Pratviel, G.; Crozet, M.D.; Wyllie, S.; Fairlamb, A.H.; Valentin, A.; Rathelot, P.; Azas, N.; Castro, B.; Verhaeghe, P. 8-Aryl-6-Chloro-3-Nitro-2-(Phenylsulfonylmethyl)Imidazo[1,2-a]Pyridines as Potent Antitrypanosomatid Molecules Bioactivated by Type 1 Nitroreductases. Eur. J. Med. Chem. 2018, 157, 115. [Google Scholar] [CrossRef]
- Nwaka, S.; Ramirez, B.; Brun, R.; Maes, L.; Douglas, F.; Ridley, R. Advancing Drug Innovation for Neglected Diseases—Criteria for Lead Progression. PLoS Negl. Trop. Dis. 2009, 3, e440. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, M.V.; Bloomer, W.D.; Rosenzweig, H.S.; O’Shea, I.P.; Wilkinson, S.R.; Kaiser, M.; Chatelain, E.; Ioset, J.-R. Discovery of potent nitrotriazole-based antitrypanosomal agents: In vitro and in vivo evaluation. Bioorg. Med. Chem. 2015, 23, 6467. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, G.A.; Spillere, A.R.; Neves, G.M.; Kagami, L.P.; von Poser, G.L.; Canto, R.F.S.; Eifler-Lima, V.R. Natural and Synthetic Coumarins as Antileishmanial Agents: A Review. Eur. J. Med. Chem. 2020, 203, 112514. [Google Scholar] [CrossRef] [PubMed]
- Faheem, F.; Dey, S.; Johri, S.; Abirami, M.; Kumar, B.K.; Taramelli, D.; Basilico, N.; Balaña-Fouce, R.; Sekhar, K.V.G.C.; Murugesan, S. Search for Structurally Diverse Heterocyclic Analogs as Dual-Acting Antimalarial and Antileishmanial Agents: An Overview. Eur. J. Med. Chem. Rep. 2022, 4, 100031. [Google Scholar] [CrossRef]
Figure 1.
Chemical structure of selenium substituted (hetero)aryl hybrids [34,35,36,37,38,39,40], used in this study.
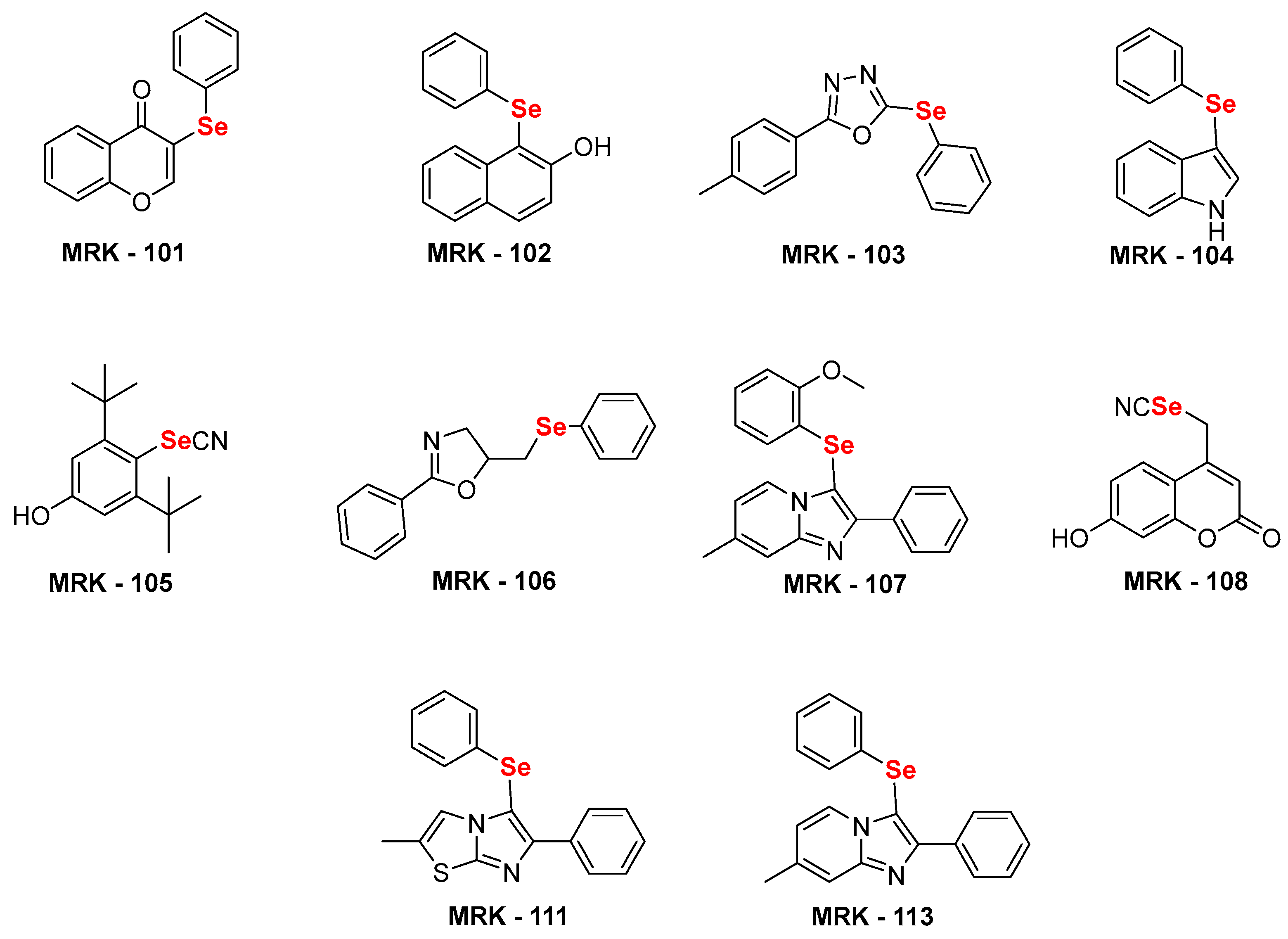
Figure 2.
Antileishmanial activity of synthesized selenium compounds MRK-105, 106, 107, 108 and 113 against promastigote (A, B, C, D and E upper graphs) and intracellular amastigotes (F, G, H, I and J lower graphs) of L. amazonensis. * and *** means p< 0,01 and p< 0,001, respectively (ANOVA and Tukey´s post test).
Figure 2.
Antileishmanial activity of synthesized selenium compounds MRK-105, 106, 107, 108 and 113 against promastigote (A, B, C, D and E upper graphs) and intracellular amastigotes (F, G, H, I and J lower graphs) of L. amazonensis. * and *** means p< 0,01 and p< 0,001, respectively (ANOVA and Tukey´s post test).
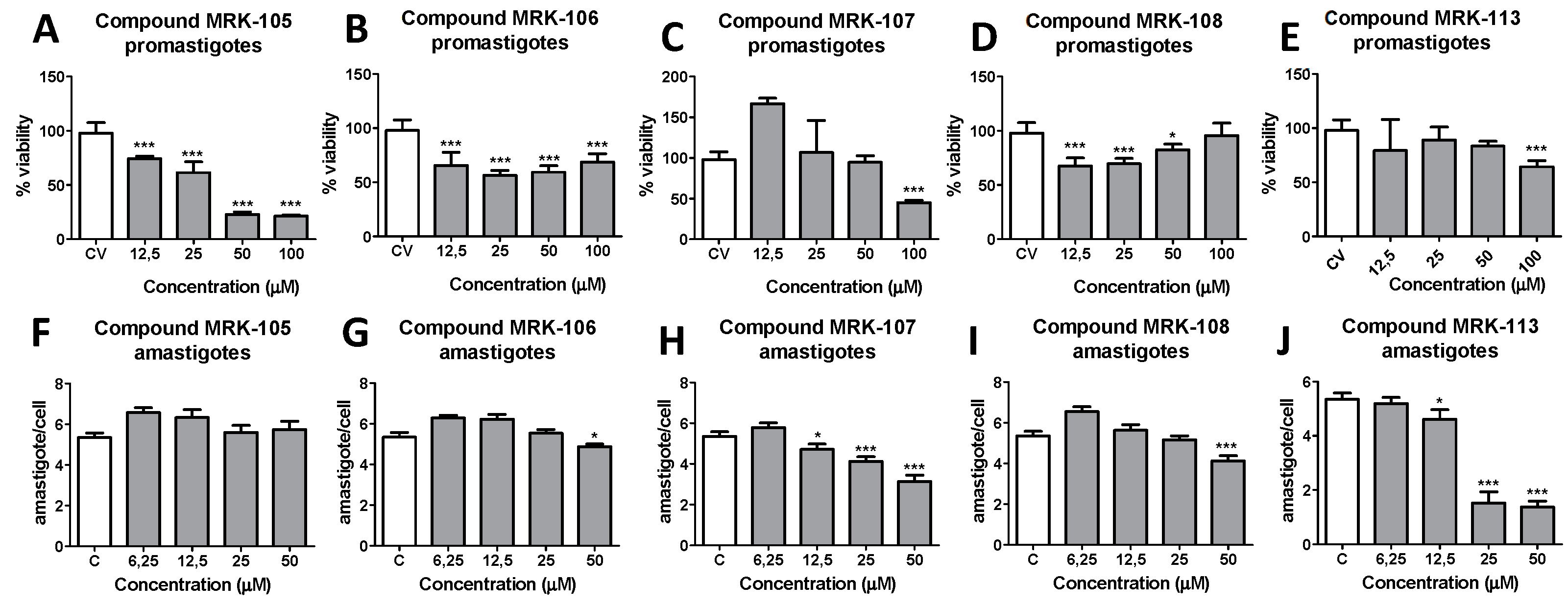

Table 1.
Antileishmanial activity of selenium compounds.
| Compound | CC50 (µM) peritoneal macrophages |
IC50 (µM) L. amazonensis promastigotes |
SIa | IC50 (µM) L. amazonensis amastigotes |
SIb |
|---|---|---|---|---|---|
| 101 | > 200 | 30.46 | 6.57 | > 50 | ind |
| 102 | > 200 | 15.15 | 13.20 | > 50 | ind |
| 103 | > 200 | 15.48 | 12.92 | > 50 | ind |
| 104 | > 200 | 16.17 | 12.37 | > 50 | ind |
| 105 | > 200 | 12.17 | 16.43 | > 50 | ind |
| 106 | > 200 | 3.95 | 50.53 | > 50 | ind |
| 107 | > 200 | 27.37 | 7.31 | 18.31 | 10.92 |
| 108 | > 200 | 4.22 | 47.31 | > 50 | ind |
| 111 | > 200 | 17.55 | 11.40 | > 50 | ind |
| 113 | > 200 | 40.98 | 4.88 | 15.93 | 12.55 |
| ANFB | 25.15 | 9.40 | 2.67 | 0.97 | 25.92 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.





MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated