
Preprint
Review
Tailored Viral-Like Particles as Drivers of Medical Breakthroughs
Altmetrics
Downloads
187
Views
78
Comments
0
A peer-reviewed article of this preprint also exists.




This version is not peer-reviewed
Submitted:
11 April 2024
Posted:
12 April 2024
You are already at the latest version
Alerts
Abstract
Despite the recognized potential of nanoparticles, only a few formulations have progressed to clinical trials, and an even smaller number have been approved by the regulatory authorities and marketed. Virus-like particles (VLPs) have emerged as promising alternatives to most explored nanoparticles because of the absence of viral genetic material, their incapacity to replicate, mimicry of viral structure, and tropism conservation. Furthermore, VLPs can be surface functionalized with small molecules to improve circulation half-life and target specificity. Through the functionalization and coating of VLPs, it is possible to optimize the response properties to a given stimulus, such as heat, pH, an alternating magnetic field, or even enzymes. Surface functionalization can also modulate other properties, such as biocompatibility, stability, and specificity, deeming VLPs as potential vaccine candidates or delivery systems. In this review, we address the different types of functionalization of VLPs, their importance, and their consequent biomedical applications.
Keywords:
Subject: Chemistry and Materials Science - Nanotechnology
1. Introduction
Nanotechnology can be defined as the design, construction, and characterization of solid colloidal materials within the nanometer range (1-100 nm) [1]. Nanoparticles (NPs) display a high surface area to volume ratio and elevated surface energy, as well as flexible magnetic, mechanical, electrical, and biological properties, rendering them suitable for a myriad of applications [1,2,3]. NPs have been explored as bioremediators [4], sensors and catalysts in environmental applications[5,6]; as drug delivery tools and imaging agents in medicine [7,8]; as pesticides and fertilizers in agriculture [9,10]; as food sensors and as electronic devices [11,12]. Among these applications, the biomedical fields are particularly relevant because NPs can potentially overcome the constraints of traditional drug delivery, namely systemic toxicity, inadequate specificity, and poor bioavailability [1,13,14]. Indeed, NPs can increase the stability and solubility of encapsulated substances, enhance the circulation time of the payload in the body, and promote the delivery of desired cargo across cellular membranes [14]. Furthermore, NPs can efficiently carry and deliver therapeutic cargoes, imaging probes, or biological substances to specific targets, paving the way for the development of more effective theranostic strategies [3,15]. However, only a few lipid- and protein-based formulations have been approved for use in clinical settings, despite the promise of NPs in general [15,16]. Each type of nanoparticle has its advantages and challenges, contributing to the expansion of the field of nanotechnology. Among the different types of nanoparticles, virus-like particles (VLPs) emerge as a versatile subclass with enormous potential to revolutionize medicine, diagnosis, and materials science [17,18,19]. Their ability to mimic viruses without causing infection makes them valuable tools for advancing research and applications in various fields.
1.1. Virus-like Particles
VLPs are typically constructed using proteins from a single virus, although structural proteins from diverse viruses may also be employed for VLP assembly [19,20]. These nanostructures were first found in the serum samples of patients with Down’s syndrome, hepatitis and leukemia, and exhibited antigenic sites at their surface [21]. VLPs range from 20 to 200 nm in size and present a set of advantageous characteristics that make them particularly useful for medical applications (Figure 1) [17,18,20,22].
VLPs can assembled with proteins a wide variety of viruses such as human papilloma virus (HPV) [24], human immunodeficiency virus (HIV) [25], Norwalk virus [26], and influenza virus [27]. Moreover, VLPs can be constructed in different expression systems, namely prokaryotic (bacteria) [28], eukaryotic (plant cells [29], insect cells [30], yeast [31] and mammalian cells [32]) and cell-free platforms [33].
To improve the efficiency, potency, and cargo loading capacity, current studies have mainly focused on understanding the self-assembly mechanisms of VLPs [34,35]. This process may be aided by different types of molecules, such as nucleic acids and supporting proteins [36].
After assembly, the surface of VLPs can be functionalized with a myriad of moieties, including ligands for T-cell receptors [37], antibody fragments [38], polysaccharides [39], polymers [40], enzymes [41], and natural amino acids [42], among others.
Although VLPs are devoid of viral genetic material, they can maintain certain features of the original virus, namely cellular receptor affinity, host cell entry points, and immunogenicity [43]. Consequently, VLPs can prompt cellular and humoral responses. Furthermore, it is possible to heighten the immune response elicited by VLPs through chemical or genetical modification of the surface of the VLP with different antigens (such as proteins or short peptides) [44]. The ability to incorporate multiple molecules onto the surface of VLPs makes them multivalent nanoplatforms that can function as vaccine formulations, imaging probes and/or therapeutic tools.
1.2. VLP Structure
There are three main classes of VLPs: enveloped, nonenveloped, and chimeric [19]. Enveloped VLPs are structurally complex, attaining a lipidic membrane from the host platform during assembly and budding and, consequently, requiring expression in eukaryotic platforms [19]. Examples of enveloped VLPs include HIV VLPs, influenza VLPs or Ebola VLPs [19]. Non-enveloped VLPs can harbor single or multiple capsid proteins and be single-, double-, or triple-layered, and can thus be produced in both prokaryotic and eukaryotic systems [19]. HPV VLPs and Norwalk VLPs are examples of non-enveloped VLPs [19]. Lastly, chimeric VLPs harness structural proteins from different viruses for assembly [19]. The classification and characteristics of some of the most studied VLPs are depicted in Table 1.
The assembly of VLPs is affected by their structure and composition. Furthermore, some factors can compromise VLP assembly, as shown in Figure 2 [36].
The presence or absence of certain natural amino acids as well as the structure acquired by the peptide chain is crucial for VLP assembly [52]. Mutagenesis of structural proteins can lead to incomplete assembly or hinder the formation of VLPs [52]. Interestingly, one study has reported that low temperatures are favorable for B19-VP2 VLP self-assembly, since they can diminish the occurrence of undesired processes, such as aggregation and protein degradation [53]. On the contrary, temperatures in the physiological range also contribute to VLP assembly [54], indicating that temperature is pivotal for VLP construction. The pH interferes with capsid-protein charge, and most studies indicate that VLP assembly is optimal at physiological pH [53,55,56]. Nonetheless, adequate VLP formation has been described for both acidic [57] and alkaline [58] pH conditions. Salts also affect protein stability through interactions with charges on the capsid protein surface, making ionic strength a key factor in VLP assembly [36]. Finally, cargo encapsulation is highly linked with VLP assembly, and cargo change and structure need to be considered when developing VLPs [36]. All these factors, together with surface functionalization, must be managed to attain functional VLPs.
1.3. VLP Downstream Processing
In order to guarantee appropriate safety and effectiveness for clinical usage, downstream processing for VLP purification is an essential step [59]. The initial step of the purification process following cell harvesting is determined by the VLPs' capacity to be discharged into the extracellular media. Cell lysis or another extraction technique could be necessary to disrupt the cells if the VLP is not released successfully, despite the fact that in certain documented cases— such as influenza VLPs generated in insect cell culture —the particles are released into the media without the need for particular precautions [60]. Generally, the strategy used is to create a cloned gene that expresses a protein with an efficient signal peptide that the secretory system will recognize and thus assist in VLP release [59,61].
A clarity phase is carried out to eliminate entire cell debris and aggregates from VLP preparations to streamline the procedures and lower the expenses of the purification process [59]. One crucial step that dramatically lowers the bulk volume and increases the ratio of VLP concentration in relation to other cellular contaminants is the capture and concentration of VLPs [59]. Depth filtration, microfiltration, tangential flow filtration (TFF), and cell sedimentation. UF/DF (Ultrafiltration/Diafiltration) and TFF with membranes or hollow fibers are often used techniques for separating VLPs from host cell contaminants, such as cell debris, digested DNA, or medium components [59]. VLPs may also be captured using the bind-and-elute method using affinity chromatography, ion-exchange chromatography (IEC), and hydrophobic interaction chromatography (HIC) [62,63].
To lower DNA and endotoxin levels, further intermediate purification processes including IEC, HIC, and ultracentrifugation are frequently needed [59].
An optional procedure used to improve the stability, homogeneity, and immunogenicity of the VLP product is disassembly and reassembly. For this phase, titration or cross-flow filtration are the selected methods [59]. All impurities left behind from processing must be eliminated in the polishing phase, which is the last stage of purification [59]. IEC, size exclusion chromatography (SEC), and UF/DF (often the crossflow technique) are frequently used for this [59,63]. The preparations are sterilized before being finalized by filtering through sterile-grade filters with a suitable pore size [63].
A downstream processing approach for VLP purification is summarized in Figure 3.
An updated list of surface functionalization of VLPs is provided in the following sections, together with information on their potential prophylactic, diagnostic and therapeutic applications.
2. Types of Surface Functionalization
Owing to their structural characteristics, VLPs can be designed by expressing proteins, peptides, nucleic acids, imaging probes, drugs, and other NPs [65,66,67] in their surface. They may also have target ligands that alter the inherent tropism of VLPs by making them specific to a particular cell, tissue, or organ [38,68]. Both chemical and genetic approaches can be employed for surface modification of VLPs [69,70] (Figure 4).
Functionalization may differ according to the nature of VLP. The functionalization of non-enveloped VLPs may be achieved through covalent conjugation to amino acid residues on the surface [71]. Cysteine, glutamic acid and lysine are natural amino acids that have been explored for functionalization [71]. For example, one study reported the conjugation of fluorescent probes to cysteine residues present on tobacco mosaic VLPs for development of photovoltaic devices [72]. Destito et al. harnessed surface lysine residues on cowpea mosaic VLPs for conjugation of folic acid to achieve efficient targeting to cancer cells [73]. However, the location of the amino acid residues within the VLP must be considered for reactivity or surface accessibility [74]. The introduction or elimination of amino acids at crucial locations using recombinant DNA technologies can be used to overcome such limitations [74]. Indeed, Thong et al. introduced a reactive cysteine on the inner surface of MS2 VLPs for incorporation of a fluorescent dye [74]. Alternatively, fusion between virus capsid proteins and the desired proteins and peptides can be performed, enabling the internal or external framing of non-enveloped VLPs [68]. This technique allows the appropriate targeting moiety to be directly attached to viral capsid proteins, despite being circumscribed to peptides and proteins [71]. The size of these peptides must also be reduced to prevent interference with VLP function [71]. For example, the epidermal growth factor was genetically fused to the C terminus of the Qβ capsid protein to form epidermal growth factor receptor specific VLPs [68]. Another study focused on HPV VLPs and depicted the fusion of green fluorescent protein (GFP) to the N or C terminus of L1 and L2 capsid proteins, to attain VLPs that can function as delivery vehicles of foreign proteins [75]. Recently, Guo et al. used genetic recombination to insert a coronavirus epitope into the Qβ capsid protein for development of a chimeric VLP vaccine [76]. Lastly, it should be noted that non-enveloped VLs can undergo non-covalent alterations by electrostatic [77,78], protein-protein [79,80], protein-nucleotide [66,81] and protein-metal [82,83] interactions. Because non-covalent modifications are frequently reversible, this might be problematic when prolonged stability is warranted.
Given the lipidic nature of their envelope, enveloped VLPs are better adapted for the display of membrane proteins [84]. This type of VLPs can also undergo pharmacological or genetic modifications, with the latter taking place when a desired protein is fused to a full-length viral protein or a transmembrane domain [85,86]. This can be achieved through loop insertion [87], N- or C- terminal modifications [87], or amino acid residue mutations [38]. For example, Boxus et al. reported the construction of several HPV chimeric VLPs through insertion of L2 epitopes into the capsid surface DE loop of HPV-16 or HPV-18 L1 proteins for development of a novel vaccine formulation against HPV [88]. Regarding N- or C-terminus modification, Gordon et al. described the fusion of the carboxy terminus of the circumsporozoite antigen derived from Plasmodium falciparum to the N-terminus of hepatitis B surface antigen, generating chimeric VLPs that were later commercialized as the Mosquirix® vaccine against malaria [87,89]. Lastly, Patel et al. employed the amino acid mutation method for construction of MS2 and Qβ VLPs with surface exposed methionine analogues comprising azide and alkyne side chains, for subsequent conjugation of azide and alkyne-harboring molecules, namely an antibody fragment and the granulocyte-macrophage colony stimulation factor [38].
A protein can undergo biochemical reactions after translation, known as post-translational modifications (PTMs), leading to changes in its properties [90,91,92]. PTMs are obtained through enzymatic binding of groups such as acetyl, glycosyl, phosphoryl, and methyl, which are added during the process [90,91,92,93]. These groups can lead to changes in protein activity and stability, physicochemical and conformational properties [90,91,92]. Consequently, PTM dysregulation leaves its mark on a wide range of illnesses. One utterly researched PTM that has been linked to cardiovascular, Alzheimer's, and cancer is phosphorylation. It has been discovered that the protein Tau, which is linked to Alzheimer's disease, is phosphorylated at forty distinct locations [94]. More significantly, there is a strong correlation between site-specific phosphorylation and the various phases of Alzheimer's disease [94]. There has been evidence linking methylation, acetylation, glycosylation, and S-nitrosylation to neurological and cardiovascular conditions [94,95]. Diabetes has been linked to dysregulated glycosylation [96]. A significant function for ubiquitination has been identified in a number of uncommon congenital illnesses, including von Hippel-Lindau syndromes [94]. The PTMs most associated with illnesses have been identified as ubiquitylation, SUMOylation, prenylation, S-palmitoylation, and glycosylation [94]. In addition, they can also improve the pharmacological features of peptides and promote their three-dimensional folding [90,91,92]. Several studies have explored the role of PTMs in VLPs [97,98]. Balieu et al. focused on the N-glycosylation pattern of the SPIKE protein within SARS-CoV-2 VLPs developed in Nicotiana benthamiana plants, and the results revealed that the overall VLP structure exhibited complex N-glycans that are similar to those expressed in mammalian platforms [97]. This highlights the potential of plants to produce novel vaccines. Another study reported that SUMO modifications are relevant for proper assembly of foot-and-mouth-disease (FMDV) VLPs, as they prevent inadequate aggregation and increase the solubility of capsid proteins [98].
3. Biomedical Applications of Functionalized VLPs
The surface functionalization of VLPs renders them flexible platforms that can be explored in different biomedical fields, such as drug delivery, vaccination, and imaging, as well as other applications, which will be presented and discussed in subsequent sections.
3.1. Drug Delivery
The precise and selected delivery of drugs to target tissues, notably anticancer drugs, is a tremendous challenge faced by medicinal chemists. One of the most auspicious strategies to surmount such limitations is to encapsulate drug molecules inside a nanoparticle that specifically targets cancer cells, which also may improve the stability of the drug in circulation [99]. Surface functionalized VLPs with active compounds may present several advantages, namely improved pharmacokinetics, optimal biodistribution, and reduced adverse effects [19,100]. Most studies that have explored VLPs as drug delivery systems have focused on microRNAs as cargo [66,101,102,103,104,105], but VLP-mediated delivery of other agents has also been described [57,66,106,107].
Pan et al. have constructed a microRNA-delivery platform grounded on bacteriophage MS2 VLPs [101]. This study aimed to investigate the potential of these nanoplatforms as delivery systems. The VLPs were constructed by fusion of pre microRNA-146a (miRNA-146a) to an MS2 cistron, which then interacted with the bacteriophage coat protein for assembly [101]. Subsequently, VLPs were conjugated to the cell penetrating peptide HIV TAT [101]. VLP concentrations in the nanomolar range were assessed both in vitro and in vivo [101]. The results showed that the VLPs successfully transferred pre-miR146a to different cells and tissues, increasing the expression of miRNA-146a both in vitro and in vivo, and subsequently suppressing its target gene [101]. This group has also focused on the effects of miR-146a on osteoclastogenesis and systemic lupus erythematosus (SLE) [102,103]. In the first study, MS2 VLPs were employed as delivery systems for miR-146a in human peripheral blood mononuclear cells (PBMCs) [102]. Addition of MS2-miR-146a-VLPs to PBMCs prompted the upregulation of miR-146a, highlighting the potential of MS2 VLPs as delivery platforms [102]. Furthermore, upon treatment with osteoclastogenesis-inducing cytokines, osteoclastogenesis-specific genes were downregulated in PBMCs transfected with MS2-miR-146a-VLPs, together with a reduced number of osteoclasts [102]. In the study, the authors investigated the role of miR-146a in systemic lupus erythematosus (SLE) and treated lupus-prone mice with MS2-miR146a VLPs [103]. Treatmnet of mice with TAT-conjugated MS2-miR146a VLPsled to increased levels of mature miR146a, which translated into a significant reduction of both autoantibody expression and proinflammatory cytokine levels [103].
Another study reported a microRNA-delivery platform based on MS2 VLPs functionalized with a GE11 polypeptide [104]. This polypeptide binds to the epidermal growth factor receptor (EGFR), which is overexpressed in several malignancies, namely hepatocellular carcinoma (HCC) [108,109]. Moreover, the maternally expressed gene 3 (MEG3), which encodes a long non-coding RNA, has shown potential as a tumor suppressor in several carcinomas, including HCC [110]. The scarcity of delivery vehicles for this long non-coding RNA spurred the study of VLPs as a valid option [104]. To package the MEG3 complementary DNA (cDNA) sequence, two mutated pac site sequences, which are viral sequences responsible for nucleic acid packaging, were inserted into MEG3 cDNA [104]. Then, the Sulfo-SMCC crosslinker was used to form a bridge between GE11 polypeptide and MS2 VLP [104]. This study demonstrated that this is a promising delivery system in cancer therapy, as it was possible to achieve swift, effective, and safe delivery of MEG3 to EGFR-positive HCC cell lines, with no activation of downstream EGFR pathways [104].
Wang et al. carried out new studies to avoid the use of a crosslinker, as it is expensive and requires preparation under strict conditions, and to increase the delivery system efficiency [105]. The authors took advantage of MS2 VLPs as delivery systems for microRNA-122 (miR-122) and placed the TAT sequence on the surface of the VLPs by phage surface display instead of using chemical crosslinkers [105]. It has been observed that miR-122 is deregulated in HCC and negatively regulates the expression of genes that partake in cancer cell proliferation, migration and invasion [111]. The authors therefore incorporated the TAT peptide into the N-terminus of the MS2 coat protein dimer and added a pac site to the pre-miR122 for packaging [105]. The results showed that these recombinant MS2 VLPs containing TAT (MS2-TAT-miR122 VLPs) are efficient, penetrating the cytomembrane and inhibiting HCC through the delivery of miR-122. This shows that this type of platform can be explored for drug delivery and as a promising treatment for HCC [105].
Interestingly, Ashley et al. reported the construction of MS2 VLPs for selective delivery of different types of moieties, namely chemotherapeutic agents, small interfering RNA (siRNA) and protein toxins to HCC [66]. To achieve selectivity, the VLPs were functionalized with an HCC-targeting peptide, SP94, through a cross-linker with a poly (ethylene glycol) (PEG) spacer [66]. Encapsulation was conducted through conjugation of a pac site to the different cargoes [66]. VLPs were thus loaded with quantum dots, three chemotherapeutic agents (doxorubicin (DOX), cisplatin and 5-fluorouracil (5-FU)), ricin toxin A-chain (RTA) and a siRNA that negatively regulates the expression of cyclin family members [66]. The results were as follows: VLPs that contained DOX, cisplatin and 5-FU were able to selectively kill HCC cells with drug concentrations under 1 nM; VLPs comprising a siRNA cocktail were able to prompt growth arrest and apoptosis of HCC cells with concentrations under 150 nM; VLPs loaded with RTA and co-functionalized with SP94 and a peptide that spurs endosomal escape were capable of killing all cells with concentrations as low as 100 fM [66]. This study highlights the flexibility of MS2 VLPs as delivery vehicles.
Another study described the attachment of DOX to VP6, a rotavirus capsid protein, for VLP assembly [57]. Lactobionic acid (LA), a ligand of asialoglycoprotein receptors (ASGPRs), is overexpressed in the membrane of HCC cells [112]. In vitro experiments demonstrated that these VLPs (DVLPLAs) were internalized by a hepatoma cell line, and most cells were killed with a concentration of 10 μg mL-1 of DOX when loaded on VLPs [57].
Kato et al. explored Rous sarcoma VLPs (RSV VLPs) as drug delivery platforms through functionalization with a hCC49 single chain fragment variable (scFv), which specifically targets the tumor-associated glycoprotein-72 (TAG-72) on the membrane of colon carcinoma cells [106]. The cytoplasmic and transmembrane domains of influenza A hemagglutinin were used to fuse the scFv to the VLPs, which were expressed in silkworm larvae [106]. Electroporation was used to encapsulate DOX, and specific targeting was assessed [106]. These results indicate that hCC49 scFv-presenting RSV VLPs can target colon carcinoma cells and efficiently deliver DOX [106].
More recently, a novel technology called SpyCatcher/SpyTag has been explored to develop targeted HBV VLPs [107]. The SpyCatcher/SpyTag system comprises a genetically encoded protein, SpyCatcher, which is designed to spontaneously covalently bind to its peptide partner SpyTag [113].
As such, VLP proteins genetically fused to SpyCatcher can be easily functionalized to moieties linked to SpyTag [113]. Hartzell et al. inserted the SpyCatcher into the c/e1 loop of HBV and tested different moieties with the SpyTag for VLP decoration [107]. For example, the authors selected the GE11 peptide, which targets EGFR, to decorate the HBV VLPs and thus specifically target inflammatory breast cancer cells [107]. Furthermore, a dimeric prodrug-activating enzyme was loaded into the VLPs, which enabled a specific uptake of the VLPs by the breast cancer cells and subsequent enzyme delivery and cell killing [107].
3.2. Vaccines
VLPs are suitable platforms for vaccination, deeming it their most common medical application [114,115]. VLP-based vaccines can provide more advantages than traditional vaccines because they structurally mimic viruses, elicit a memory response, and have several epitopes in their structure, which prompt humoral and cell-mediated responses, enhancing immunity [114,115]. Furthermore, since VLPs do not have a viral genome, they cannot infect or replicate, thus not compromising safety [114,115]. Several VLP-based vaccines against viruses have been developed in recent years and are now commercially available (Table 2).
VLPs can function as stand-alone vaccines against their native viruses, relying on their virus-like structure to elicit an immune response [114,115]. Notwithstanding, VLP functionalization has also been described for the development of vaccination formulations, namely against cancer. An example of the application of surface functionalized VLPs in vaccines is the VLPs evaluated by Patel and Swartz [38], which derive from bacteria-infecting viruses (bacteriophages MS2 and Qβ). This bestows them with an important competitive edge over VLPs derived from human-infecting viruses, such as HIV, because they are less susceptible to pre-existing antibodies in the human population, potentially preserving the immunogenicity of these vaccines. These VLPs were produced with methionine analogs that contain azide (azidohomoalanine: AHA) and alkyne (homopropargylglycine: HPG) terminal groups, which are non-natural amino acids (nnAAs) and are exposed at the surface, allowing for conjugation of several biomolecules, namely nucleic acids, proteins and small molecules, such as PEG [38]. These biomolecules were functionalized with azide and alkyne for conjugation to VLPs, which occurred in a single step catalyzed by Cu(I) using click chemistry. Consequently, VLP functionalization with three different ligands was achieved in one step. This study depicted VLP production through direct conjugation with antigens from idiotype antibody fragments, cytokine GM-CSF and the immunostimulatory CpG DNA, to produce new candidate tumor idiotype vaccines for the treatment of B-cell lymphoma [38].
VLPs have also been modified with the human epidermal growth factor receptor 2 (HER2). Palladini et al. harnessed the SpyCatcher/SpyTag system to construct multivalent HER2 extracellular domain (ECD)-presenting AP205 VLPs [128]. The immunogenic and therapeutic potential of this vaccine formulation was assessed in mice bearing HER2-overexpressing mammary carcinoma cells and compared with a traditional DNA vaccine [128]. This vaccine was able to elicit strong anti-HER2 autoantibody responses, limit the spontaneous development of HER2-overexpressing carcinomas and hamper the growth of HER2-overexpressing tumors introduced into wild-type mice [128]. The VLP vaccine showed better results than the DNA vaccine, and no significant side effects were observed in the treated mice, which corroborates the potential of VLPs as cancer vaccines [128].
Salazar-González et al. designed B19 human parvovirus VLPs with chimeric VP2 proteins and functionalized them with two insulin-like growth factor-1 receptor (IGF-1R) epitopes through fusion with the N-terminus of VP2 [129]. IGF-1R has been related to angiogenesis in different malignancies, and epitopes P8, a B-cell epitope within the extracellular domain of the receptor, and 249, a T-cell receptor that binds to both major histocompatibility complexes, have been identified as relevant epitopes, and were thus selected for the development of VLP vaccines [129]. Several chimeric multiepitope VLPs were thus generated and were employed in a prophylactic setting of 4 weekly immunizations preceding 4T1 cell inoculation in mice [129]. Then, 4 weekly intraperitoneal injections VLPs were administered, and this regime was able to protect mice from tumor formation and growth following cell inoculation [129]. It was possible to observe specific antibodies against the presented epitopes, but no significant T-cell responses, indicating that further studies are required [129].
The recent COVID-19 pandemic has also spurred the development of novel vaccination strategies, and one study described the fusion of the SARS-CoV-2 S protein to the Gag protein of either HIV-1 or simian immunodeficiency virus (SIV) in a mRNA vaccine for assembly of chimeric VLPs [130]. This vaccine was inoculated into mice and its effects in an in vivo model were assessed [130]. A high level of S-trimer-binding and neutralizing antibodies was observed, demonstrating that this platform can be explored as a vaccine against different pathologic agents [130].
3.3. Imaging
Imaging is pivotal for diagnosis and visualization of disease location and progression, providing greater monitoring of the disease in the patient and thus leading to a more successful treatment [131,132]. Given the high biocompatibility and easy surface functionalization of VLPs, their combination with several imaging agents has been described [18,131].
Li et al. modified selectively the surface-coating protein P8 of the bacteriophage M13 on the side chains of lysine, aspartic or glutamic acid and tyrosine residues for conjugation [133]. After surface modification, the bacteriophage M13 undergoes a double modification with fluorescein dyes (reactive fluorescein N-hydroxy succinimidyl ester: FL-NHS) and folic acid (FA) [133]. A first incubation with FL-NHS was carried out to give rise to a VLP dye/phage (M13-FL), followed by dialysis and then M13-FL was functionalized with folate-azide using a tyrosine conjugation method [133]. The results obtained by Li et al. show that M13 was double-modified (FL-M13-FA), and through three-dimensional imaging, it was possible to observe that the FL-M13-FA VLPs were in the cytoplasm of KB cells and could be explored as fluorescent probes for imaging in this type of human cells [133].
Another example is the work conducted by Sun et al., which focused on the development of simian virus 40 (SV40)-based VLPs carrying near-infrared quantum dots for imaging and peptides for targeting atherosclerotic plaques [134]. Atherosclerosis is a systemic pathology that affects arteries and is characterized by the deposition of plaques in arterial walls, which may spawn myocardial infarction and stroke [135]. Traditionally, the diagnosis of this disease was only carried out in later stages, but novel imaging modalities, namely optical imaging, are currently under study [136]. Within optical imaging, near-infrared quantum dots emerge as promising candidates, due to their detection sensitivity in deep tissues [137]. Sun and colleagues genetically inserted an atherosclerosis-targeting peptide into the HI loop region of the SV40 major capsid protein VP1, and a thrombin inhibitor peptide (Hirulog) into the N-terminus of VP1 [134]. The authors also developed VLPs harboring both peptide that targets fibrin at an advanced stage of atherosclerosis and Hirulog, and VLPs that only contained a peptide that targets vascular cell adhesion molecule-1 (VCAM-1), a marker for the original state of atherosclerosis [134]. These engineered proteins underwent self-assembly together with near-infrared quantum dots to form multifunctional VLPs, which were then evaluated in an in vivo model [134]. The multifunctional VLPs were intravenously injected into mice, and the quantum dots were able to elicit sufficient signal for imaging of atherosclerotic plaques in mice, and the targeting moieties allowed for specificity to the aorta, namely in sites where atherosclerotic plaques are typically formed [134]. Immunohistochemistry analysis revealed that the targeted nanoplatforms colocalized with their targets, and Hirulog was able to exert its function and treat thrombin[134]. All these data underline the functionality and practicality of VLPs as theranostic platforms [134].
A VLP for PET imaging was designed upon conjugation of an antibody against EGFR to MS2 capsid proteins and consequently labeled with copper-64 (64Cu) [138]. To construct this tailored nanoplatform, an oxidative coupling strategy of aminophenols and anilines was employed [139]. Anilines were incorporated into MS2 viral capsids via introduction of p-aminophenylalanine on the outer surface, and aminophenol was inserted into the antibody through non-site-specific lysine modification with nitrophenol-NHS-ester [139]. The two were then coupled through an oxidative reaction [139]. A maleimide-chelator was then added to the VLPs for radiolabeling with 64Cu, and the resulting VLPs were evaluated both in vitro and in vivo [138]. In vitro studies revealed specific binding to the EGFR receptor present on the membrane of different cancer cell lines [138]. The radiolabeled EGFR-targeting VLPs were injected into mice bearing tumor xenografts and ex vivo PET imaging was performed to assess VLP location [138]. Moderate tumor uptake and long circulation periods were observed, but no significant specific uptake was observed, indicating that further improvements are required to achieve a targeting VLP that is useful for PET imaging [138].
More recently, physalis mottle virus (PhMV) VLPs were loaded with the fluorescent dye Cy5.5 and gadolinium (Gd)(III) complexes for near-infrared fluorescence and MRI, respectively, and functionalized with a targeting peptide [140]. Thiol-maleimide chemistry was used to conjugate the DOTA chelator (and subsequent radiolabeling with Gd(III)) and Cy5.5 to the inner surface of the VLPs. In addition, amine-N-hydroxysuccinimide ester chemistry was used to conjugate the DGEA peptide, which targets integrin α2β1, a receptor that is overexpressed in the prostate cancer cell line PC-3 [140]. PC-3 cells were treated modified VLPs, and it was possible to observe a high fluorescent signal and subsequent high VLP cell uptake [140]. VLPs were injected into mice harboring PC-3 tumors, and imaging analysis revealed that the VLPs were efficiently taken up by the tumor [140].
3.4. Biosensing
Biosensing is based on receptor-transducer devices capable of transforming a biological signal into an electrical response and consequently originating a measurable signal [141]. These instruments typically comprise an analyte, a bioreceptor, a transducer, electronics and a display unit, and are utilized to measure pH levels, pathogen concentrations, toxic chemicals, among others [141]. VLPs, besides vaccination, imaging, and drug delivery, have also been recently explored for biosensing.
Mieloch et al. developed biomimetic VLPs with superparamagnetic iron oxide nanoparticles and the receptor binding domain (RBD) of SARS-CoV-2 [142]. The bioactivity of the particles was assessed, and it was possible to observe that the VLPs were capable of binding to SARS-CoV-2 specific targets, such as angiotensin-converting enzyme 2 (ACE2) and toll-like receptor 4 (TLR4) [142]. Furthermore, these particles were capable of capturing SARS-CoV-2 targeting antibodies with high specificity, and are therefore presented as an alternative to typical COVID-19 detection kits [142]. However, this data is preliminary, and further tests on the ability to immobilize these particles on detection structures are still required [142].
Among the different biosensors under development for disease diagnosis, nucleic acid-based fluorescent biosensors have gained relevance, in particular for living sample analysis [143]. Within these biosensors, the hybridization chain reaction (HCR), an enzyme-free isothermal amplification method, is highlighted due to its potential in living imaging [144]. The main principle of HCR is based on assembly of stable DNA monomers following exposure to a target DNA sequences [145]. Basically, two stable DNA hairpins are placed in solution, and the introduction of initiator sequences spurs hybridization that results in double strands, whose molecular weight in inversely proportional to the initiator concentration [145]. HCR can be conjugated to aptamer triggers and therefore function as an amplifying transducer in biosensors [145].Traditional HCR, however, is not highly sensitive due to its single linear extension circuit and cannot be easily taken up by biological systems due to the negative charge of nucleic acids. Chen et al. attempted to circumvent these issues by designing a novel strategy that harnesses VLPs [146]. First, a stack design was employed to develop a hierarchical HCR (H-HCR) that can graft multiunit HCR products [146]. The elements of this H-HCR were then packaged into VP3 (structural protein of the human bocavirus 1 capsid) VLPs engineered to present arginine-glycine-aspartic acid (RGD) peptides, which targets the αvβ3 integrin (a cancer biomarker) [146]. Afterwards, a multibranched DNA scaffold was designed to bear the H-HCR components and heighten the H-HCR produced signals [146]. Additionally, a photocleavage linker (PC-linker) was introduced to bestow a spatiotemporal light-powered behaviour, and thus ensure that imaging signals are not lost during delivery [146]. The authors designated this method as a genetically engineered VLP-armoured and multibranched DNA-scaffold-corbelled H-HCR, and tested it for the detection of a cancer-associated mRNA marker (miRNA-155) in vitro, as well as for imaging in living cells [146]. This method proved to be highly specific and ultrasensitive, and is ancapable of providing strong imaging in both healthy and cancerfous cells, and well as in tumour-bearing mice [146]. This innovative strategy introduces novel paths to achieve stronger and more robust disease diagnosis.
Another study reported the design of a coulometric enzyme immunoassay for detection of immunoglobulin M (IgM) against Hantaan viruses (HTNVs) by exploring VLPs as detecting moieties [147]. First, the authors functionalized screen-printed carbon electrodes (SPCEs), which are widely used electrodes in point-of-care biosensors, with paste-exfoliated graphene (a carbon material presenting good electrical conductivity) bearing a carboxylic acid group and a thionine mediator [147]. Then, an antibody against HTNV IgM was immobilized onto the SPCE surface [147]. HTNV VLPs were employed as antigens for second recognition of anti-HTNV IgM antibodies, and these VLPs were functionalized with horseradish peroxidase for coulometric measurement of hydrogen peroxide reduction mediated by thionine following application of a potential [147]. This system could detect anti-HTNV IgM in human serum with high selectivity and sensitivity, and highlights the potential of VLPs in biosensing, especially in disease marker detection [147].
4. Challenges of VLP-Based Approaches
Despite the promising results exhibited by VLPs, it is important to highlight that only a few VLP-based formulations have been approved and commercialized. These formulations were exclusively approved for vaccination, which is linked to the high immunogenicity of VLPs. This can severely hamper their use as imaging and/or drug delivery tools. Furthermore, there are several hurdles related to VLP and expression host selection, functionalization, scalability, downstream processing, and stability. All these factors must be taken into consideration when developing novel VLP-based tools.
4.1. Selection of VLP Type and Expression Platform
Most of the marketed VLPs are non-enveloped, due to their simplicity. Production of non-enveloped single-protein capsid VLPs is typically performed in Escherichia coli or yeast, and both systems allow for high production yields and easy scale up [148]. Furthermore, yeast expression systems can provide PTMs, which can play a pivotal role in VLP function [148]. Interestingly, due to their simplicity, these VLPs can be produced in cell-free environment. Non-enveloped multiple-capsid protein VLPs, on the other hand, are typically produced in the baculovirus-insect expression system [148]. This system can also provide high yields and PTMs, but it is not as easily scalable and regulated as E.coli and yeast, and requires more complex purification processes [148]. Production in microbial cells typically entails cell lysis, which can release cell-derived impurities and consequently affect the downstream processing [149]. Moreover, the production process of non-enveloped VLPs typically generates non-uniform particles [150]. To overcome these hurdles, these VLPs typically undergo in vitro disassembly and reassembly, which has proved to improve stability [151] and immunogenicity [152]. The main advantages of non-enveloped VLPs include their well-characterized nature and their stability, given that they are less sensitive to external conditions (purification, temperature or sheer force) than enveloped VLPs [153]. This justifies that most FDA-approved VLPs are non-enveloped. Nonetheless, they are not as suitable for the presentation of membrane proteins as enveloped VLPs, due to the origin of their envelope [84]. Even so, the advantages presented by these VLPs justify that most VLP-based platforms developed in recent years for drug delivery and imaging are non-enveloped.
Enveloped VLPs are structurally more complex than non-enveloped VLPs, since they attain their lipidic envelope from their expression platform during assembly and budding [19]. Indeed, this type of VLP can present one or several glycoproteins on their lipidic membrane, which function as target antigens and can therefore elicit an immune response and prompt the production of neutralizing antibodies [19,154]. For this reason, production of enveloped VLPs requires eukaryotic expression platforms [19]. The major issue with enveloped VLPs is their poor biophysical characterization given that they are not as structurally uniform as non-enveloped VLPs [148,153]. Moreover, as abovementioned, enveloped VLPs are more sensitive to external conditions than non-enveloped VLPs, which means their immunogenicity, integrity and stability can be more easily compromised [153]. Enveloped VLPs also require correct folding and glycosylation for adequate function as vaccine formulations [153]. These challenges deem enveloped VLPs more difficult to work with and consequently to implement in the clinical setting, but strategies such as computational design, genetic engineering, in vitro assembly and novel processing and purification methods can be explored to circumvent these limitations [153].
Regarding expression hosts, each platform has its advantages and disadvantages, which are depicted in Table 3.
4.2. VLP Preparation
VLP production typically entails three steps: production (or upstream processing), purification (or downstream processing), and formulation (for VLP-based vaccines) [19,20]. Production starts with cloning of the viral genes that encode the proteins of interest, followed by expression and assembly in the expression systems [19,20]. The produced VLPs are then transferred to the solution through cell lysis [19,20]. Clarification is performed to eliminate most aggregates and cell contaminants [19,20]. Further purification is typically required to attain integral VLPs, followed by a concentration step, and a final polishing step is done to remove any remaining residual nucleic acids and proteins [19,20]. Formulation typically entails the incorporation of adjuvants and extra ingredients to obtain a safe and efficient vaccine [19,20].
Besides selection of the most adequate expression system, it is also critical to select the most suitable culture mode, since it affects concentration, productivity, and yield [153,155]. There are three different culture modes for VLP production: continuous cultivation, batch and fed batch [153,155]. Continuous cultivation typically ensures the highest productivity, since it relies on cultures in a log phase, and therefore allows to obtain continuous production [153,155]. This culture mode has been reported for VLP production in different expression platforms, such as insect cells [156] and mammalian cells [157]. The main advantages of this culture mode include a short turn-around period, reduced number of production steps and continuous downstream processing [153,155]. Within continuous production, there are different methods: chemostat, morbidostat, stressostat and turbidostat [153,155]. Among these, chemostat is the most frequently used mode, as it permits to modulate growth rates and product formation. Notwithstanding, this method is also associated with a high contamination risk and low product concentrations [153,155]. In the batch mode, all the required nutrients are added only at the start of the culture process, and this brings a low contamination risk [153,155]. The main drawbacks of such a method include low viral titers, low productivity, temperature sensitivity and product and substrate inhibition [153,155]. Substrate inhibition can be overcome by diminishing the lag phase, which is what occurs the fed-batch mode [153,155]. In this method, the exponential growth phase can be augmented by selective incorporation of nutrients, and it is possible to increase the period of culture to attain higher cell densities [153,155]. Nonetheless, scale-up is costly since there is an inefficient use of culture medium [153,155]. Factor such as cost, cell growth, yield, contamination, particle size, cell lysis, among others, need to be considered when selecting the culture mode [153,155]. It is also important to note that these culture modes were only tested to produce VLP-based vaccines. The studies that report the design and evaluation of VLPs for other applications, namely imaging and drug delivery, are carried out at a small scale, and do not account for scale up. Studies focused on the production process of VLPs for such applications are thus required, to determine whether it is possible to attain such VLPs with high stability and productivity, for further implementation in the medical field.
Regarding VLP assembly, different factors are at play. Single or multi-capsid proteins must assemble in expression platforms or cell-free systems, and this assembly may rely on interactions with scaffolding proteins and/or nucleic acids [36,153]. For example, it has been reported that adeno-associated virus (AAV) relies on the assembly-activating protein (AAP) for VLP assembly [58]. Another example is HIV-1 Gag VLPs, which utilize RNA as a scaffold for assembly [158]. Such molecules must be carefully considered during VLP assembly because they can severely affect VLP applications by increasing their immunogenicity [36]. Moreover, factors such as ionic strength, pH, temperature, and stoichiometry heavily influence VLP assembly, and so optimal conditions must be fine-tuned to avoid VLP malformation [36,153].
Downstream processing is a critical step in VLP preparation, as it allows to separate a myriad of proteins from the desired VLPs [36,153]. When VLPs are attained through transduction, viral vectors are used to deliver genetic material to cells, which originates proteins of similar size and molecular weight and consequently makes the separation process more burdensome [153]. Transfection, on the other hand, does not rely on viral vectors, and thus facilitates separation [153]. Serum supplements possess a high protein content, which can hamper purification, and so the utilization of serum-free media may also be beneficial [153]. Regarding cell lysis, as previously mentioned, this step is only necessary for non-secreting expression systems, as certain insect and mammalian cells secrete VLPs into the cell supernatants [20]. For non-secreting platforms, treatment with detergent-bearing solutions is sufficient for most eukaryotic cells, but other platforms, such as bacterial, yeast and plant cells, possess a cell wall and therefore require mechanical treatment for VLP release, which could hamper their integrity [20]. Ultracentrifugation is typically used to achieve purity, but this process is laborious, is not scalable and increases the risk of VLP aggregation [20]. Chromatography methods are thus utilized to circumvent these limitations, but these processes may require additional steps in VLP processing, which can also be a disadvantage [20]. Additionally, precautions should be taken to ensure that the formulation of VLP-based vaccines is of high quality [159]. Lastly, conditions such as ion and salt concentrations, as well as pH, must be meticulously controlled during each downstream processing step, as they greatly impact VLP aggregation [20].
After downstream processing, VLPs must be characterized to ensure quality and consistency [20,148]. Before characterization, ammonium sulfate or PEG are employed to precipitate high molecular weight VLPs and therefore eliminate low molecular weight impurities [20]. It is critical to ensure precipitate purity before moving on to downstream analysis, since precipitation can prompt VLP disassembly [20,153]. Biochemical characterization techniques to detect the structural proteins of VLPs include enzyme-linked immunosorbent assay (ELISA), sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and western blot (WB) [20,153]. Notwithstanding, through these techniques it is not possible to distinguish between integral, partially assembled or unassembled VLPs [20]. Furthermore, these methods display low sensitivity, demand high volumes and are time-consuming, which impacts downstream studies [153]. Techniques such as high-performance liquid chromatography (HPLC), SDS-capillary gel electrophoresis and matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) have emerged as alternatives to the classical biochemical characterization methods. Regarding biophysical characterizations, several methods can be explored: transmission electron microscopy, to detect particle size and polydispersity; atomic force microscopy (AFM), to determine VLP morphology; dynamic light scattering (DLS) to assess particle aggregation, among others[153]. Sample preparation for some of these techniques involves drying, which can introduce artifacts into the samples [153]. Cryo-EM can be used to overcome this handicap, as it employs VLPs in solution, but it is only feasible for sufficiently homogenous samples [153]. Lastly, rigorous measurement of particle size and quantification is necessary for correct VLP dosing, and it is highly demanding [153]. Approaches such as nanoparticle track analysis (NTA) or electrospray-differential mobility analysis (ES-DMA) can be used to accurately keep track of batch-to-batch consistency, which is particularly useful for VLP-based vaccine formulations [153].
4.3. Functionalization
As previously stated, the ability to functionalize VLPs on their inner and/or outer surface is one of the major selling points of these nanostructures [19]. The selected functionalization method can also influence VLP production and function. For instance, genetic fusion is simpler than chemical functionalization and can broaden the applications of VLPs, but it is also associated with low protein yields or inappropriate folding [20,160].Chemical conjugation does not have such limitations, but it is more expensive and laborious, and typically leads to a stronger immune response, which can be a disadvantage in drug delivery and imaging [20,160]. It is also important to consider the number of amino acids that are introduced, and hydrophobicity, given that these factors are pivotal for adequate VLP structure and function [20,160].
4.4. Immunogenicity
One of the main features of VLPs is their capacity to mimic the virus from which they derive and therefore present an icosahedral structure bearing multiple epitopes that activate antigen-presenting cells (APCs) [19,131,154]. VLPs bind to pattern recognition receptors (PRRs) present on the surface of dendritic cells and are consequently taken up for processing [19,131,154]. VLP-derived peptides are then presented on the surface by MHC class I and class II molecules, together with co-stimulatory molecules, for presentation to cytotoxic T and T helper cells, respectively [19,131,154]. Cytotoxic T cells are responsible for a cellular immune response, whereas T helper cells activate both T and B cells, and the latter in turn produce antibodies and are responsible for the humoral immune response [19,131,154]. Interestingly, VLPs are capable of binding directly to B cells and thus provoking humoral responses without requiring the intervention of APCs [19,131,154]. All in all, VLPs are excellent substrates to elicit both cellular and humoral immune responses [19,131,154]. This is an ideal scenario when the end-goal is to develop VLP-based vaccines. This becomes a hurdle when VLPs are explored as nanocarriers of imaging probes and/or therapeutic drugs. In vitro studies in cultured cells do not account for immunogenicity, as it does not affect VLP function, but this is a severe hindrance when we consider an in vivo scenario. Tests to assess the immunogenicity of VLPs, together with strategies to reduce it are thus required, and should be explored by researchers. Opting for VLPs derived from viruses with low antigenicity and employing engineering techniques to modify VLPs or replace their surface epitopes are among promising approaches to reduce VLP immunogenicity [161].
5. Emerging Approaches and Future Perspectives
VLP applications conjugate different biological fields, namely immunology, virology, microbiology, and vaccinology. Scientists have been drawn to the use of VLPs as special tools to develop novel strategies and methodologies due to their qualities. For instance, with the use of designer endonucleases, genome editing technology allows humans to alter a target organism's genome in order to add or remove particular DNA fragments from a cell [162]. The targeting technique used in early gene editing, homologous recombination, was incredibly ineffective and prone to off-target effects [163]. The paradigm was changed by the discovery of designer endonucleases. Three main types of designer endonucleases are frequently used for DNA modification: clustered regularly interspaced short palindromic repeats/CRISPR-associated (CRISPR/Cas), transcription activator-like effector nuclease (TALEN) and zinc finger nuclease (ZNF) and [164].
VLPs have thus also been explored as delivery vehicle of genome editing enzymes [165,166,167,168,169]. Long-term expression of genome editing enzymes has been associated with cytotoxicity, which prompted authors to develop VLPs that can transiently deliver the CRISPR/Cas9 system. Knopp et al. to encapsulate CRISPR/Cas in “all-in-one” non-integrating particles of the murine leukemia virus through transfection of SpCas9 and single guide RNA (sgRNA)-encoding plasmids [168]. Then, transient delivery of these RNAs was performed in multiple murine and human cell lines [168]. Nevertheless, the authors observed that knockout efficiencies were lower than those observed for constitutively active integrating viral vectors [168]. Notwithstanding, stable SpCas9 expression led to cell arrest and reduced cell metabolic activity and growth, which highlights the advantages of transient delivery [168]. Other investigations have also focused on transient delivery of the CRISPR/Cas technology in VLPs and have shown promising results [165,166,167]. VLP-mediated delivery of other genome editing enzymes has also been described. Cai et al. described the use of HIV-1 Gag particles for delivery of ZNFs or TALENs by co-packaging the designer endonucleases and donor RNA in “all-in-one“particles. This translated into homology-directed repair and reduced off-target effects [169]. It is important to note that these studies did not report VLP functionalization to achieve delivery, which is something that should be tested to determine whether it is possible to perform tailored genome editing that only affects specific cells by displaying targeting moieties on the VLP surface.
Another emerging approach in the field of nanomedicine is artificial intelligence (AI), which has risen as a potent tool to predict and analyse interactions between different molecules, predict optimal dosage, drug combination and administration window for each patient, and ultimately aid in the design and implementation of novel nanomedicines. [170]. Li et al. used computer assistance to create a few novel enzymes and achieve catalytic reactions not seen in nature [171]. After redesigning the Bacillus aspartic acid enzyme, several synthetic product-amino acid synthetases that were stereoselective and had perfect position selectivity were produced [171]. Cunningham et al. introduced a customised machine-learning technique, Hierarchical statistical mechanical modelling (HSM) [172]. HSM can reliably predict the affinities of PBD–peptide interactions across a variety of protein families [172]. BANDIT is a Bayesian machine-learning technique for molecule-target predictions, created by Madhukar et al., which ca predict drug binding targets [173]. Ianevski et al. has applied DECREASE, a machine learning model, to assess the effects of combining different medicines while minimizing the number of dose-response measurements, and this has the potential to considerably lower the cost of medication combination screening [174]. Gainza et al. established a conceptual framework based on a geometric deep learning algorithm, MaSIF (molecular surface interaction fingerprinting) to capture fingerprints relevant to biomolecular interactions [175]. This method allowed to predict protein-molecule interactions using chemical and geometric signatures on the surface of molecules [175]. All these AI methods, together with many others that are currently under development, have a lot of room to grow and might significantly influence basic studies on the composition and properties of proteins, and revolutionize nanomedicine.
Even with AI's help, the prediction and design of biomolecular interactions remains difficult despite the recent advancements. For instance, gathering pre-existing data and building a database thereafter takes time. Due to the vast number of proteins and their diverse structures, compiling current information and building a database using a straightforward approach is difficult. After this, more work and verification are still required to use AI to obtain a generalised design scheme based on the data that is already available. Furthermore, it is currently difficult to consider the impact of these variables in the AI-assisted design process since a variety of factors, including the microenvironment and the architecture of the proteins themselves, influence how well proteins operate.
6. Conclusions
Nanotechnology, and in particular nanomedicine, has emerged in recent years as a field aimed at addressing the gaps that still exist in traditional medicine, namely in the treatment of diseases such as cancer [176]. Conventional treatments are typically associated with off-target effects and limited delivery to the desired target [176]. Targeted therapy has emerged as a means to tackle these limitations, given that it provides specific delivery to the desired site, minimizing side-effects; allows for the delivery of higher doses; and can reach tissues that are typically difficult to access [177]. Among the different delivery vehicles, nanoparticles are of particular interest, due to their ability to improve drug solubility and stability, as well as effectively package and delivery different cargoes [3]. Notwithstanding, only a few NP-based formulations (inorganic, liposomal and polymeric particles) have been commercialized [15,16]. These NPs typically rely on passive targeting and undergo clearance by dendritic cells and phagocytes [15]. Surface modification with PEG has been explored to extend the circulation time of NPs and diminish their uptake by the reticuloendothelial system [15]. The problem is that PEGylation can also increase immunogenicity and reduce target uptake due to the recognition of PEG by the immune system [178]. Within the different types of nanoplatforms, VLPs are a particularly promising alternative to conventional NPs due to their capacity for genetic and chemical modification on their exterior and interior surfaces via amino acid chemistries and self-assembly procedures, which offer precise targeting, solubility, and stability. They are regarded as potent platforms to present antigens, epitopes, polymers, and ligands in a dense repeating array to enhance therapeutic effectiveness. They are also regarded as fitting packaging and delivery platforms for anticancer drugs and genetic materials. For this reason, VLPs have been modified on the surface to achieve targeting and subsequently explored as drug delivery vehicles of moieties such as miRNA [66,101,102,103,104,105] and chemotherapeutic agents [57,66,106,107]. Functionalized VLPs stand out for their increased specificity as nanocarriers for drug delivery. Furthermore, the applicability of VLPs in vaccine formulations has shown that they are considered safe and capable of eliciting a strong immune response against infectious and non-infectious pathologies, namely viral diseases [116,117,118,119,120,121,122,123,124,125,126,127,130] and cancer [38,128,129]. VLPs are also able to bear imaging agents, and have been explored as diagnostic agents for optical imaging [133,134,140], MRI [140] and PET imaging [138]. More recently, VLPs have also been explored in biosensing for disease marker detection [146]. Technologies such as genome editing, and artificial intelligence could also be explored in the design and production of novel VLP-based formulation. Brought together, these data indicate that VLPs are highly versatile platforms for a wide variety of biomedical applications. The described show that the field has matured from its early stages. But there are still obstacles in the way of developing the area of viral medication delivery. The most evident obstacle is whether the viral carrier would be immune to pre-existing or de novo acquired adaptive immunity, and if this would result in side effects or lower efficacy following repeated delivery [51]. Nonetheless, several aspects need to be considered when designing targeting VLPs, especially their immunogenicity, stability and ability to efficiently reach their target in vivo. Immunogenicity is particularly troubling for drug delivery and imaging, but surface passivation and immune modification techniques have showed promise in lowering the immune response elicited by VLPs [179]. Furthermore, hurdles such as high production costs, scalability and regulatory limitations hamper the implementation of VLPs in the biomedical setting [153]. Despite these limitations, the potential exhibited by functionalized VLPs far outweighs the presented hindrances, which deems further studies, particularly focused on scalable production and immunogenicity, necessary, in the hopes of moving towards in vivo tests and, in the end, an increasing number of marketed VLPs.
Author Contributions
R.T. was involved in methodology, validation, investigation, and writing—original draft preparation. S.M. was involved in validation, data curation, formal analysis, and writing—original draft preparation. A.F. was involved in writing—original draft preparation. J.D.G.C. was responsible for conceptualization, investigation, writing—review and editing and funding. R.M. was responsible for conceptualization, methodology, validation, formal analysis, investigation, data curation, writing—original draft preparation, writing—review and editing, visualization, supervision, and project administration. All authors have read and agreed to the published version of the manuscript.
Funding
S.M. was funded by Fundação para a Ciência e Tecnologia (FCT) (UI/BD/154474/2022). R.T., R.M. and J.D.G.C. were funded by FCT through projects UID/Multi/04349/ 2020 and PTDC/QUI-OUT/3854/2021
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chenthamara, D.; Subramaniam, S.; Ramakrishnan, S.G.; Krishnaswamy, S.; Essa, M.M.; Lin, F.H.; Qoronfleh, M.W. Therapeutic Efficacy of Nanoparticles and Routes of Administration. Biomater Res 2019, 23, 20. [CrossRef]
- Kim, B.Y.; Rutka, J.T.; Chan, W.C. Nanomedicine. N Engl J Med 2010, 363, 2434-2443. [CrossRef]
- Chen, G.; Roy, I.; Yang, C.; Prasad, P.N. Nanochemistry and Nanomedicine for Nanoparticle-based Diagnostics and Therapy. Chem Rev 2016, 116, 2826-2885. [CrossRef]
- Chaudhary, P.; Ahamad, L.; Chaudhary, A.; Kumar, G.; Chen, W.-J.; Chen, S. Nanoparticle-Mediated Bioremediation as a Powerful Weapon in the Removal of Environmental Pollutants. Journal of Environmental Chemical Engineering 2023, 109591.
- Willner, M.R.; Vikesland, P.J. Nanomaterial Enabled Sensors for Environmental Contaminants. J Nanobiotechnology 2018, 16, 95. [CrossRef]
- Gómez-López, P.; Puente-Santiago, A.; Castro-Beltrán, A.; do Nascimento, L.A.S.; Balu, A.M.; Luque, R.; Alvarado-Beltrán, C.G. Nanomaterials and Catalysis for Green Chemistry. Current Opinion in Green and Sustainable Chemistry 2020, 24, 48-55.
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano Based Drug Delivery Systems: Recent Developments and Future Prospects. J Nanobiotechnology 2018, 16, 71. [CrossRef]
- Han, X.; Xu, K.; Taratula, O.; Farsad, K. Applications of Nanoparticles in Biomedical Imaging. Nanoscale 2019, 11, 799-819. [CrossRef]
- Chaud, M.; Souto, E.B.; Zielinska, A.; Severino, P.; Batain, F.; Oliveira-Junior, J.; Alves, T. Nanopesticides in Agriculture: Benefits and Challenge in Agricultural Productivity, Toxicological Risks to Human Health and Environment. Toxics 2021, 9. [CrossRef]
- Mejias, J.; Salazar, F.; Pérez Amaro, L.; Hube, S.; Rodriguez, M.; Alfaro, M. Nanofertilizers: A Cutting-Edge Approach to Increase Nitrogen Use Efficiency in Grasslands. Frontiers in Environmental Science 2021, 9, 52.
- Ghosh, T.; Raj, G.B.; Dash, K.K. A Comprehensive Review on Nanotechnology Based Sensors for Monitoring Quality and Shelf Life of Food Products. Measurement: Food 2022, 100049.
- Payal; Pandey, P. Role of Nanotechnology in Electronics: A Review of Recent Developments and Patents. Recent Pat Nanotechnol 2022, 16, 45-66. [CrossRef]
- McNamara, K.; Tofail, S.A. Nanoparticles in Biomedical Applications. Advances in Physics: X 2017, 2, 54-88.
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering Precision Nanoparticles for Drug Delivery. Nat Rev Drug Discov 2021, 20, 101-124. [CrossRef]
- Shan, X.; Gong, X.; Li, J.; Wen, J.; Li, Y.; Zhang, Z. Current Approaches of Nanomedicines in the Market and Various Stage of Clinical Translation. Acta Pharm Sin B 2022, 12, 3028-3048. [CrossRef]
- Namiot, E.D.; Sokolov, A.V.; Chubarev, V.N.; Tarasov, V.V.; Schioth, H.B. Nanoparticles in Clinical Trials: Analysis of Clinical Trials, FDA Approvals and Use for COVID-19 Vaccines. Int J Mol Sci 2023, 24. [CrossRef]
- Rohovie, M.J.; Nagasawa, M.; Swartz, J.R. Virus-Like Particles: Next-Generation Nanoparticles for Targeted Therapeutic Delivery. Bioeng Transl Med 2017, 2, 43-57. [CrossRef]
- Benjamin, C.; Brohlin, O.; Shahrivarkevishahi, A.; Gassensmith, J.J. Virus Like Particles: Fundamental Concepts, Biological Interactions, and Clinical Applications. Nanoparticles for Biomedical Applications 2020, 153-174.
- Nooraei, S.; Bahrulolum, H.; Hoseini, Z.S.; Katalani, C.; Hajizade, A.; Easton, A.J.; Ahmadian, G. Virus-Like Particles: Preparation, Immunogenicity and Their Roles as Nanovaccines and Drug Nanocarriers. J Nanobiotechnology 2021, 19, 59. [CrossRef]
- Zeltins, A. Construction and Characterization of Virus-Like Particles: A Review. Mol Biotechnol 2013, 53, 92-107. [CrossRef]
- Bayer, M.E.; Blumberg, B.S.; Werner, B. Particles Associated with Australia Antigen in the Sera of Patients with Leukaemia, Down's Syndrome and Hepatitis. Nature 1968, 218, 1057-1059. [CrossRef]
- Lee, L.A.; Niu, Z.; Wang, Q. Viruses and Virus-Like Protein Assemblies—Chemically Programmable Nanoscale Building Blocks. Nano Research 2009, 2, 349-364.
- Mejia-Mendez, J.L.; Vazquez-Duhalt, R.; Hernandez, L.R.; Sanchez-Arreola, E.; Bach, H. Virus-Like Particles: Fundamentals and Biomedical Applications. Int J Mol Sci 2022, 23. [CrossRef]
- Wang, J.W.; Roden, R.B. Virus-Like Particles for the Prevention of Human Papillomavirus-Associated Malignancies. Expert Rev Vaccines 2013, 12, 129-141. [CrossRef]
- Martins, S.A.; Santos, J.; Silva, R.D.; Rosa, C.; Cabo Verde, S.; Galamba Correia, J.D.; Melo, R. How Promising Are HIV-1-Based Virus-Like Particles for Medical Applications? Frontiers in Cellular and Infection Microbiology 2022, 1526. [CrossRef]
- Herbst-Kralovetz, M.; Mason, H.S.; Chen, Q. Norwalk Virus-Like Particles as Vaccines. Expert Rev Vaccines 2010, 9, 299-307. [CrossRef]
- Pushko, P.; Tretyakova, I. Influenza Virus Like Particles (VLPs): Opportunities for H7N9 Vaccine Development. Viruses 2020, 12. [CrossRef]
- Huang, X.; Wang, X.; Zhang, J.; Xia, N.; Zhao, Q. Escherichia coli-Derived Virus-Like Particles in Vaccine Development. NPJ Vaccines 2017, 2, 3. [CrossRef]
- Marsian, J.; Lomonossoff, G.P. Molecular Pharming - VLPs Made in Plants. Curr Opin Biotechnol 2016, 37, 201-206. [CrossRef]
- Gopal, R.; Schneemann, A. Production and Application of Insect Virus-Based VLPs. Methods Mol Biol 2018, 1776, 125-141. [CrossRef]
- Srivastava, V.; Nand, K.N.; Ahmad, A.; Kumar, R. Yeast-Based Virus-like Particles as an Emerging Platform for Vaccine Development and Delivery. Vaccines (Basel) 2023, 11. [CrossRef]
- Pulix, M.; Lukashchuk, V.; Smith, D.C.; Dickson, A.J. Molecular Characterization of HEK293 Cells as Emerging Versatile Cell Factories. Curr Opin Biotechnol 2021, 71, 18-24. [CrossRef]
- Rodriguez-Limas, W.A.; Sekar, K.; Tyo, K.E. Virus-Like Particles: The Future of Microbial Factories and Cell-Free Systems as Platforms for Vaccine Development. Curr Opin Biotechnol 2013, 24, 1089-1093. [CrossRef]
- Wang, Y.; Douglas, T. Bioinspired Approaches to Self-Assembly of Virus-like Particles: From Molecules to Materials. Acc Chem Res 2022, 55, 1349-1359. [CrossRef]
- Ikwuagwu, B.; Hartman, E.; Mills, C.E.; Tullman-Ercek, D. Systematic Engineering of Virus-Like Particles to Identify Self-Assembly Rules for Shifting Particle Size. Virology 2023, 579, 137-147. [CrossRef]
- Le, D.T.; Muller, K.M. In Vitro Assembly of Virus-Like Particles and Their Applications. Life (Basel) 2021, 11. [CrossRef]
- Derdak, S.V.; Kueng, H.J.; Leb, V.M.; Neunkirchner, A.; Schmetterer, K.G.; Bielek, E.; Majdic, O.; Knapp, W.; Seed, B.; Pickl, W.F. Direct Stimulation of T Lymphocytes by Immunosomes: Virus-Like Particles Decorated with T Cell Receptor/CD3 Ligands Plus Costimulatory Molecules. Proc Natl Acad Sci U S A 2006, 103, 13144-13149. [CrossRef]
- Patel, K.G.; Swartz, J.R. Surface Functionalization of Virus-Like Particles by Direct Conjugation Using Azide-Alkyne Click Chemistry. Bioconjug Chem 2011, 22, 376-387. [CrossRef]
- Li, X.; Pan, C.; Sun, P.; Peng, Z.; Feng, E.; Wu, J.; Wang, H.; Zhu, L. Orthogonal Modular Biosynthesis of Nanoscale Conjugate Vaccines for Vaccination Against Infection. Nano Res 2022, 15, 1645-1653. [CrossRef]
- Kraj, P.; Selivanovitch, E.; Lee, B.; Douglas, T. Polymer Coatings on Virus-like Particle Nanoreactors at Low Ionic Strength-Charge Reversal and Substrate Access. Biomacromolecules 2021, 22, 2107-2118. [CrossRef]
- Cayetano-Cruz, M.; Coffeen, C.F.; Valadez-Garcia, J.; Montiel, C.; Bustos-Jaimes, I. Decoration of Virus-Like Particles with an Enzymatic Activity of Biomedical Interest. Virus Res 2018, 255, 1-9. [CrossRef]
- Carrico, Z.M.; Farkas, M.E.; Zhou, Y.; Hsiao, S.C.; Marks, J.D.; Chokhawala, H.; Clark, D.S.; Francis, M.B. N-Terminal Labeling of Filamentous Phage to Create Cancer Marker Imaging Agents. ACS Nano 2012, 6, 6675-6680. [CrossRef]
- Mohsen, M.O.; Zha, L.; Cabral-Miranda, G.; Bachmann, M.F. Major Findings and Recent Advances in Virus-Like Particle (VLP)-Based Vaccines. Semin Immunol 2017, 34, 123-132. [CrossRef]
- Naskalska, A.; Pyrc, K. Virus Like Particles as Immunogens and Universal Nanocarriers. Pol J Microbiol 2015, 64, 3-13.
- Durous, L.; Rosa-Calatrava, M.; Petiot, E. Advances in Influenza Virus-Like Particles Bioprocesses. Expert Rev Vaccines 2019, 18, 1285-1300. [CrossRef]
- Marzi, A.; Feldmann, H. Ebola Virus Vaccines: An Overview of Current Approaches. Expert Rev Vaccines 2014, 13, 521-531. [CrossRef]
- Shang, W.; Liu, J.; Yang, J.; Hu, Z.; Rao, X. Dengue Virus-Like Particles: Construction and Application. Appl Microbiol Biotechnol 2012, 94, 39-46. [CrossRef]
- Pumpens, P.; Grens, E. The True Story and Advantages of the Famous Hepatitis B Virus Core Particles: Outlook 2016. Mol Biol (Mosk) 2016, 50, 558-576. [CrossRef]
- Fu, Y.; Li, J. A Novel Delivery Platform Based on Bacteriophage MS2 Virus-Like Particles. Virus Res 2016, 211, 9-16. [CrossRef]
- Essus, V.A.; Souza Junior, G.S.E.; Nunes, G.H.P.; Oliveira, J.D.S.; de Faria, B.M.; Romao, L.F.; Cortines, J.R. Bacteriophage P22 Capsid as a Pluripotent Nanotechnology Tool. Viruses 2023, 15. [CrossRef]
- Schwarz, B.; Douglas, T. Development of Virus-Like Particles for Diagnostic and Prophylactic Biomedical Applications. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2015, 7, 722-735. [CrossRef]
- Palucha, A.; Loniewska, A.; Satheshkumar, S.; Boguszewska-Chachulska, A.M.; Umashankar, M.; Milner, M.; Haenni, A.L.; Savithri, H.S. Virus-Like Particles: Models for Assembly Studies and Foreign Epitope Carriers. Prog Nucleic Acid Res Mol Biol 2005, 80, 135-168. [CrossRef]
- Sanchez-Rodriguez, S.P.; Munch-Anguiano, L.; Echeverria, O.; Vazquez-Nin, G.; Mora-Pale, M.; Dordick, J.S.; Bustos-Jaimes, I. Human Parvovirus B19 Virus-Like Particles: In Vitro Assembly and Stability. Biochimie 2012, 94, 870-878. [CrossRef]
- Jaballah, S.A.; Bailey, G.D.; Desfosses, A.; Hyun, J.; Mitra, A.K.; Kingston, R.L. In Vitro Assembly of the Rous Sarcoma Virus Capsid Protein Into Hexamer Tubes at Physiological Temperature. Sci Rep 2017, 7, 2913. [CrossRef]
- Xu, J.; Guo, H.C.; Wei, Y.Q.; Dong, H.; Han, S.C.; Ao, D.; Sun, D.H.; Wang, H.M.; Cao, S.Z.; Sun, S.Q. Self-Assembly of Virus-Like Particles of Canine Parvovirus Capsid Protein Expressed from Escherichia coli and Application as Virus-Like Particle Vaccine. Appl Microbiol Biotechnol 2014, 98, 3529-3538. [CrossRef]
- Barklis, E.; Alfadhli, A.; McQuaw, C.; Yalamuri, S.; Still, A.; Barklis, R.L.; Kukull, B.; Lopez, C.S. Characterization of the In Vitro HIV-1 Capsid Assembly Pathway. J Mol Biol 2009, 387, 376-389. [CrossRef]
- Zhao, Q.; Chen, W.; Chen, Y.; Zhang, L.; Zhang, J.; Zhang, Z. Self-Assembled Virus-Like Particles From Rotavirus Structural Protein VP6 for Targeted Drug Delivery. Bioconjug Chem 2011, 22, 346-352. [CrossRef]
- Le, D.T.; Radukic, M.T.; Muller, K.M. Adeno-Associated Virus Capsid Protein Expression in Escherichia coli and Chemically Defined Capsid Assembly. Sci Rep 2019, 9, 18631. [CrossRef]
- Vicente, T.; Roldao, A.; Peixoto, C.; Carrondo, M.J.; Alves, P.M. Large-Scale Production and Purification of VLP-Based Vaccines. J Invertebr Pathol 2011, 107 Suppl, S42-48. [CrossRef]
- Lai, C.C.; Cheng, Y.C.; Chen, P.W.; Lin, T.H.; Tzeng, T.T.; Lu, C.C.; Lee, M.S.; Hu, A.Y. Process Development for Pandemic Influenza VLP Vaccine Production Using a Baculovirus Expression System. J Biol Eng 2019, 13, 78. [CrossRef]
- Hillebrandt, N.; Vormittag, P.; Bluthardt, N.; Dietrich, A.; Hubbuch, J. Integrated Process for Capture and Purification of Virus-Like Particles: Enhancing Process Performance by Cross-Flow Filtration. Front Bioeng Biotechnol 2020, 8, 489. [CrossRef]
- Pniewski, T.; Kapusta, J.; Bociag, P.; Wojciechowicz, J.; Kostrzak, A.; Gdula, M.; Fedorowicz-Stronska, O.; Wojcik, P.; Otta, H.; Samardakiewicz, S.; et al. Low-Dose Oral Immunization with Lyophilized Tissue of Herbicide-Resistant Lettuce Expressing Hepatitis B Surface Antigen for Prototype Plant-Derived Vaccine Tablet Formulation. J Appl Genet 2011, 52, 125-136. [CrossRef]
- Hutchins, B.; Sajjadi, N.; Seaver, S.; Shepherd, A.; Bauer, S.R.; Simek, S.; Carson, K.; Aguilar-Cordova, E. Working Toward an Adenoviral Vector Testing Standard. Mol Ther 2000, 2, 532-534. [CrossRef]
- Peixoto, C.; Sousa, M.F.; Silva, A.C.; Carrondo, M.J.; Alves, P.M. Downstream Processing of Triple Layered Rotavirus Like Particles. J Biotechnol 2007, 127, 452-461. [CrossRef]
- Kaczmarczyk, S.J.; Sitaraman, K.; Young, H.A.; Hughes, S.H.; Chatterjee, D.K. Protein Delivery Using Engineered Virus-Like Particles. Proc Natl Acad Sci U S A 2011, 108, 16998-17003. [CrossRef]
- Ashley, C.E.; Carnes, E.C.; Phillips, G.K.; Durfee, P.N.; Buley, M.D.; Lino, C.A.; Padilla, D.P.; Phillips, B.; Carter, M.B.; Willman, C.L.; et al. Cell-Specific Delivery of Diverse Cargos by Bacteriophage MS2 Virus-Like Particles. ACS Nano 2011, 5, 5729-5745. [CrossRef]
- Li, F.; Zhang, Z.P.; Peng, J.; Cui, Z.Q.; Pang, D.W.; Li, K.; Wei, H.P.; Zhou, Y.F.; Wen, J.K.; Zhang, X.E. Imaging Viral Behavior in Mammalian Cells with Self-Assembled Capsid-Quantum-Dot Hybrid Particles. Small 2009, 5, 718-726. [CrossRef]
- Pokorski, J.K.; Hovlid, M.L.; Finn, M.G. Cell Targeting with Hybrid Qβ Virus-Like Particles Displaying Epidermal Growth Factor. Chembiochem 2011, 12, 2441-2447. [CrossRef]
- Bustos-Jaimes, I.; Soto-Roman, R.A.; Gutierrez-Landa, I.A.; Valadez-Garcia, J.; Segovia-Trinidad, C.L. Construction of Protein-Functionalized Virus-Like Particles of Parvovirus B19. J Biotechnol 2017, 263, 55-63. [CrossRef]
- Zackova Suchanova, J.; Neburkova, J.; Spanielova, H.; Forstova, J.; Cigler, P. Retargeting Polyomavirus-Like Particles to Cancer Cells by Chemical Modification of Capsid Surface. Bioconjug Chem 2017, 28, 307-313. [CrossRef]
- Smith, M.T.; Hawes, A.K.; Bundy, B.C. Reengineering Viruses and Virus-Like Particles Through Chemical Functionalization Strategies. Curr Opin Biotechnol 2013, 24, 620-626. [CrossRef]
- Miller, R.A.; Presley, A.D.; Francis, M.B. Self-Assembling Light-Harvesting Systems from Synthetically Modified Tobacco Mosaic Virus Coat Proteins. J Am Chem Soc 2007, 129, 3104-3109. [CrossRef]
- Destito, G.; Yeh, R.; Rae, C.S.; Finn, M.G.; Manchester, M. Folic Acid-Mediated Targeting of Cowpea Mosaic Virus Particles to Tumor Cells. Chem Biol 2007, 14, 1152-1162. [CrossRef]
- Tong, G.J.; Hsiao, S.C.; Carrico, Z.M.; Francis, M.B. Viral Capsid DNA Aptamer Conjugates as Multivalent Cell-Targeting Vehicles. J Am Chem Soc 2009, 131, 11174-11178. [CrossRef]
- Windram, O.P.; Weber, B.; Jaffer, M.A.; Rybicki, E.P.; Shepherd, D.N.; Varsani, A. An Investigation Into the Use of Human Papillomavirus Type 16 Virus-Like Particles as a Delivery Vector System for Foreign Proteins: N- and C-Terminal Fusion of GFP to the L1 and L2 Capsid Proteins. Arch Virol 2008, 153, 585-589. [CrossRef]
- Guo, Y.; Guo, R.; Ma, Y.; Chang, W.; Ming, S.; Yang, G.; Guo, Y. Chimeric Virus-Like Particles of Universal Antigen Epitopes of Coronavirus and Phage Qβ Coat Protein Trigger the Production of Neutralizing Antibodies. Curr Top Med Chem 2021, 21, 1235-1250. [CrossRef]
- Kostiainen, M.A.; Hiekkataipale, P.; Jose, Á.; Nolte, R.J.; Cornelissen, J.J. Electrostatic Self-Assembly of Virus–Polymer Complexes. Journal of materials chemistry 2011, 21, 2112-2117.
- Ng, B.C.; Chan, S.T.; Lin, J.; Tolbert, S.H. Using Polymer Conformation to Control Architecture in Semiconducting Polymer/Viral Capsid Assemblies. ACS Nano 2011, 5, 7730-7738. [CrossRef]
- Parent, K.N.; Deedas, C.T.; Egelman, E.H.; Casjens, S.R.; Baker, T.S.; Teschke, C.M. Stepwise Molecular Display Utilizing Icosahedral and Helical Complexes of Phage Coat and Decoration Proteins in the Development of Robust Nanoscale Display Vehicles. Biomaterials 2012, 33, 5628-5637. [CrossRef]
- Patterson, D.P.; Schwarz, B.; El-Boubbou, K.; van der Oost, J.; Prevelige, P.E.; Douglas, T. Virus-Like Particle Nanoreactors: Programmed Encapsulation of the Thermostable CelB Glycosidase Inside the P22 Capsid. Soft Matter 2012, 8, 10158-10166.
- Galaway, F.A.; Stockley, P.G. MS2 Virus-Like Particles: A Robust, Semisynthetic Targeted Drug Delivery Platform. Mol Pharm 2013, 10, 59-68. [CrossRef]
- Musick, M.A.; McConnell, K.I.; Lue, J.K.; Wei, F.; Chen, C.; Suh, J. Reprogramming Virus Nanoparticles to Bind Metal Ions Upon Activation with Heat. Biomacromolecules 2011, 12, 2153-2158. [CrossRef]
- Minten, I.J.; Wilke, K.D.; Hendriks, L.J.; van Hest, J.C.; Nolte, R.J.; Cornelissen, J.J. Metal-Ion-Induced Formation and Stabilization of Protein Cages Based on the Cowpea Chlorotic Mottle Virus. Small 2011, 7, 911-919. [CrossRef]
- Grgacic, E.V.; Anderson, D.A. Virus-Like Particles: Passport to Immune Recognition. Methods 2006, 40, 60-65. [CrossRef]
- Carvalho, S.B.; Freire, J.M.; Moleirinho, M.G.; Monteiro, F.; Gaspar, D.; Castanho, M.; Carrondo, M.J.T.; Alves, P.M.; Bernardes, G.J.L.; Peixoto, C. Bioorthogonal Strategy for Bioprocessing of Specific-Site-Functionalized Enveloped Influenza-Virus-Like Particles. Bioconjug Chem 2016, 27, 2386-2399. [CrossRef]
- Martins, S.A.; Santos, J.; Cabo Verde, S.; Correia, J.D.G.; Melo, R. Construction of HER2-Specific HIV-1-Based VLPs. Bioengineering (Basel) 2022, 9. [CrossRef]
- Gordon, D.M.; McGovern, T.W.; Krzych, U.; Cohen, J.C.; Schneider, I.; LaChance, R.; Heppner, D.G.; Yuan, G.; Hollingdale, M.; Slaoui, M.; et al. Safety, Immunogenicity, and Efficacy of a Recombinantly Produced Plasmodium falciparum circumsporozoite Protein-Hepatitis B Surface Antigen Subunit Vaccine. J Infect Dis 1995, 171, 1576-1585. [CrossRef]
- Boxus, M.; Fochesato, M.; Miseur, A.; Mertens, E.; Dendouga, N.; Brendle, S.; Balogh, K.K.; Christensen, N.D.; Giannini, S.L. Broad Cross-Protection Is Induced in Preclinical Models by a Human Papillomavirus Vaccine Composed of L1/L2 Chimeric Virus-Like Particles. J Virol 2016, 90, 6314-6325. [CrossRef]
- Laurens, M.B. RTS,S/AS01 Vaccine (MosquirixTM): An Overview. Hum Vaccin Immunother 2020, 16, 480-489. [CrossRef]
- Ramazi, S.; Zahiri, J. Posttranslational Modifications in Proteins: Resources, Tools and Prediction Methods. Database (Oxford) 2021, 2021. [CrossRef]
- Farley, A.R.; Link, A.J. Identification and Quantification of Protein Posttranslational Modifications. Methods Enzymol 2009, 463, 725-763. [CrossRef]
- Muller, M.M. Post-Translational Modifications of Protein Backbones: Unique Functions, Mechanisms, and Challenges. Biochemistry 2018, 57, 177-185. [CrossRef]
- Ojha, R.; Prajapati, V.K. Cognizance of Posttranslational Modifications in Vaccines: A Way to Enhanced Immunogenicity. J Cell Physiol 2021, 236, 8020-8034. [CrossRef]
- Vellosillo, P.; Minguez, P. A Global Map of Associations Between Types of Protein Posttranslational Modifications and Human Genetic Diseases. iScience 2021, 24, 102917. [CrossRef]
- Liu, Y.P.; Zhang, T.N.; Wen, R.; Liu, C.F.; Yang, N. Role of Posttranslational Modifications of Proteins in Cardiovascular Disease. Oxid Med Cell Longev 2022, 2022, 3137329. [CrossRef]
- Vanuopadath, M.; Nair, D.; Gopalakrishnan Nair, B.; Sadasivan Nair, S. Post-Translational Modifications of Proteins: Biomarkers and Therapeutic Targets for Diabetes Related Complications. Current Proteomics 2016, 13, 251-270.
- Balieu, J.; Jung, J.W.; Chan, P.; Lomonossoff, G.P.; Lerouge, P.; Bardor, M. Investigation of the N-Glycosylation of the SARS-CoV-2 S Protein Contained in VLPs Produced in Nicotiana benthamiana. Molecules 2022, 27. [CrossRef]
- Lee, C.D.; Yan, Y.P.; Liang, S.M.; Wang, T.F. Production of FMDV Virus-Like pPrticles by a SUMO Fusion Protein Approach in Escherichia coli. J Biomed Sci 2009, 16, 69. [CrossRef]
- Ikwuagwu, B.; Tullman-Ercek, D. Virus-Like Particles for Drug Delivery: A Review of Methods and Applications. Curr Opin Biotechnol 2022, 78, 102785. [CrossRef]
- Yuan, B.; Liu, Y.; Lv, M.; Sui, Y.; Hou, S.; Yang, T.; Belhadj, Z.; Zhou, Y.; Chang, N.; Ren, Y.; et al. Virus-Like Particle-Based Nanocarriers as an Emerging Platform for Drug Delivery. J Drug Target 2023, 31, 433-455. [CrossRef]
- Pan, Y.; Zhang, Y.; Jia, T.; Zhang, K.; Li, J.; Wang, L. Development of a MicroRNA Delivery System Based on Bacteriophage MS2 Virus-Like Particles. FEBS J 2012, 279, 1198-1208. [CrossRef]
- Yao, Y.; Jia, T.; Pan, Y.; Gou, H.; Li, Y.; Sun, Y.; Zhang, R.; Zhang, K.; Lin, G.; Xie, J.; et al. Using a Novel MicroRNA Delivery System to Inhibit Osteoclastogenesis. Int J Mol Sci 2015, 16, 8337-8350. [CrossRef]
- Pan, Y.; Jia, T.; Zhang, Y.; Zhang, K.; Zhang, R.; Li, J.; Wang, L. MS2 VLP-Based Delivery of MicroRNA-146a Inhibits Autoantibody Production in Lupus-Prone Mice. Int J Nanomedicine 2012, 7, 5957-5967. [CrossRef]
- Chang, L.; Wang, G.; Jia, T.; Zhang, L.; Li, Y.; Han, Y.; Zhang, K.; Lin, G.; Zhang, R.; Li, J.; et al. Armored Long Non-Coding RNA MEG3 Targeting EGFR Based on Recombinant MS2 Bacteriophage Virus-Like Particles Against Hepatocellular Carcinoma. Oncotarget 2016, 7, 23988-24004. [CrossRef]
- Wang, G.; Jia, T.; Xu, X.; Chang, L.; Zhang, R.; Fu, Y.; Li, Y.; Yang, X.; Zhang, K.; Lin, G.; et al. Novel miR-122 Delivery System Based on MS2 Virus Like Particle Surface Displaying Cell-Penetrating Peptide TAT for Hepatocellular Carcinoma. Oncotarget 2016, 7, 59402-59416. [CrossRef]
- Kato, T.; Yui, M.; Deo, V.K.; Park, E.Y. Development of Rous sarcoma Virus-Like Particles Displaying hCC49 scFv for Specific Targeted Drug Delivery to Human Colon Carcinoma Cells. Pharm Res 2015, 32, 3699-3707. [CrossRef]
- Hartzell, E.J.; Lieser, R.M.; Sullivan, M.O.; Chen, W. Modular Hepatitis B Virus-like Particle Platform for Biosensing and Drug Delivery. ACS Nano 2020, 14, 12642-12651. [CrossRef]
- Li, Z.; Zhao, R.; Wu, X.; Sun, Y.; Yao, M.; Li, J.; Xu, Y.; Gu, J. Identification and Characterization of a Novel Peptide Ligand of Epidermal Growth Factor Receptor for Targeted Delivery of Therapeutics. FASEB J 2005, 19, 1978-1985. [CrossRef]
- Berasain, C.; Avila, M.A. The EGFR Signalling System in the Liver: From Hepatoprotection to Hepatocarcinogenesis. J Gastroenterol 2014, 49, 9-23. [CrossRef]
- Braconi, C.; Kogure, T.; Valeri, N.; Huang, N.; Nuovo, G.; Costinean, S.; Negrini, M.; Miotto, E.; Croce, C.M.; Patel, T. MicroRNA-29 Can Regulate Expression of the Long Non-Coding RNA Gene MEG3 in Hepatocellular Cancer. Oncogene 2011, 30, 4750-4756. [CrossRef]
- Nakao, K.; Miyaaki, H.; Ichikawa, T. Antitumor Function of microRNA-122 Against Hepatocellular Carcinoma. J Gastroenterol 2014, 49, 589-593. [CrossRef]
- D'Souza, A.A.; Devarajan, P.V. Asialoglycoprotein Receptor Mediated Hepatocyte Targeting - Strategies and Applications. J Control Release 2015, 203, 126-139. [CrossRef]
- Brune, K.D.; Leneghan, D.B.; Brian, I.J.; Ishizuka, A.S.; Bachmann, M.F.; Draper, S.J.; Biswas, S.; Howarth, M. Plug-and-Display: Decoration of Virus-Like Particles Via Isopeptide Bonds for Modular Immunization. Sci Rep 2016, 6, 19234. [CrossRef]
- Kheirvari, M.; Liu, H.; Tumban, E. Virus-Like Particle Vaccines and Platforms for Vaccine Development. Viruses 2023, 15. [CrossRef]
- Kushnir, N.; Streatfield, S.J.; Yusibov, V. Virus-Like Particles as a Highly Efficient Vaccine Platform: Diversity of Targets and Production Systems and Advances in Clinical Development. Vaccine 2012, 31, 58-83. [CrossRef]
- Keating, G.M.; Noble, S. Recombinant Hepatitis B Vaccine (Engerix-B®): A Review of Its Immunogenicity and Protective Efficacy Against Hepatitis B. Drugs 2003, 63, 1021-1051. [CrossRef]
- Hussain, Z.; Ali, S.S.; Husain, S.A.; Raish, M.; Sharma, D.R.; Kar, P. Evaluation of Immunogenicity and Reactogenicity of Recombinant DNA Hepatitis B Vaccine Produced in India. World J Gastroenterol 2005, 11, 7165-7168. [CrossRef]
- Tele, S.A.; Martins, R.M.; Lopes, C.L.; dos Santos Carneiro, M.A.; Souza, K.P.; Yoshida, C.F. Immunogenicity of a Recombinant Hepatitis B Vaccine (Euvax-B) in Haemodialysis Patients and Staff. Eur J Epidemiol 2001, 17, 145-149. [CrossRef]
- Shivananda; Somani, V.; Srikanth, B.S.; Mohan, M.; Kulkarni, P.S. Comparison of Two Hepatitis B Vaccines (GeneVac-B and Engerix-B) in Healthy Infants in India. Clin Vaccine Immunol 2006, 13, 661-664. [CrossRef]
- Batdelger, D.; Dandii, D.; Dahgwahdorj, Y.; Erdenetsogt, E.; Oyunbileg, J.; Tsend, N.; Bayarmagnai, B.; Jirathitikal, V.; Bourinbaiar, A.S. Clinical Experience with Therapeutic Vaccines Designed for Patients with Hepatitis. Curr Pharm Des 2009, 15, 1159-1171. [CrossRef]
- Hieu, N.T.; Kim, K.H.; Janowicz, Z.; Timmermans, I. Comparative Efficacy, Safety and Immunogenicity of Hepavax-Gene and Engerix-B, Recombinant Hepatitis B Vaccines, in Infants Born to HBsAg and HBeAg Positive Mothers in Vietnam: An Assessment at 2 Years. Vaccine 2002, 20, 1803-1808. [CrossRef]
- Venters, C.; Graham, W.; Cassidy, W. Recombivax-HB: Perspectives Past, Present and Future. Expert Rev Vaccines 2004, 3, 119-129. [CrossRef]
- Lakshmi, G.; Reddy, R.P.; Kumar, K.K.; Bhavani, N.V.; Dayanand, M. Study of the Safety, Immunogenicity and Seroconversion of a Hepatitis-B Vaccine in Malnourished Children of India. Vaccine 2000, 18, 2009-2014. [CrossRef]
- Abraham, P.; Mistry, F.P.; Bapat, M.R.; Sharma, G.; Reddy, G.R.; Prasad, K.S.; Ramanna, V. Evaluation of a New Recombinant DNA Hepatitis B Vaccine (Shanvac-B). Vaccine 1999, 17, 1125-1129. [CrossRef]
- Shi, L.; Sings, H.L.; Bryan, J.T.; Wang, B.; Wang, Y.; Mach, H.; Kosinski, M.; Washabaugh, M.W.; Sitrin, R.; Barr, E. GARDASIL®: Prophylactic Human Papillomavirus Vaccine Development—From Bench Top to Bed-Side. Clin Pharmacol Ther 2007, 81, 259-264. [CrossRef]
- Monie, A.; Hung, C.F.; Roden, R.; Wu, T.C. Cervarix™: A Vaccine for the Prevention of HPV 16, 18-Associated Cervical Cancer. Biologics 2008, 2, 97-105.
- Herzog, C.; Hartmann, K.; Kunzi, V.; Kursteiner, O.; Mischler, R.; Lazar, H.; Gluck, R. Eleven Years of Inflexal V® - A Virosomal Adjuvanted Influenza Vaccine. Vaccine 2009, 27, 4381-4387. [CrossRef]
- Palladini, A.; Thrane, S.; Janitzek, C.M.; Pihl, J.; Clemmensen, S.B.; de Jongh, W.A.; Clausen, T.M.; Nicoletti, G.; Landuzzi, L.; Penichet, M.L.; et al. Virus-Like Particle Display of HER2 Induces Potent Anti-Cancer Responses. Oncoimmunology 2018, 7, e1408749. [CrossRef]
- Salazar-Gonzalez, J.A.; Ruiz-Cruz, A.A.; Bustos-Jaimes, I.; Moreno-Fierros, L. Expression of Breast Cancer-Related Epitopes Targeting the IGF-1 Receptor in Chimeric Human Parvovirus B19 Virus-Like Particles. Mol Biotechnol 2019, 61, 742-753. [CrossRef]
- Zhang, P.; Falcone, S.; Tsybovsky, Y.; Singh, M.; Gopan, V.; Miao, H.; Seo, Y.; Rogers, D.; Renzi, I.; Lai, Y.T.; et al. Increased Neutralization Potency and Breadth Elicited by a SARS-CoV-2 mRNA Vaccine Forming Virus-Like Particles. Proc Natl Acad Sci U S A 2023, 120, e2305896120. [CrossRef]
- Chung, Y.H.; Cai, H.; Steinmetz, N.F. Viral Nanoparticles for Drug Delivery, Imaging, Immunotherapy, and Theranostic Applications. Adv Drug Deliv Rev 2020, 156, 214-235. [CrossRef]
- Sun, X.; Cui, Z. Virus-Like Particles as Theranostic Platforms. Advanced Therapeutics 2020, 3, 1900194.
- Li, K.; Chen, Y.; Li, S.; Nguyen, H.G.; Niu, Z.; You, S.; Mello, C.M.; Lu, X.; Wang, Q. Chemical Modification of M13 Bacteriophage and Its Application in Cancer Cell Imaging. Bioconjug Chem 2010, 21, 1369-1377. [CrossRef]
- Sun, X.; Li, W.; Zhang, X.; Qi, M.; Zhang, Z.; Zhang, X.E.; Cui, Z. In Vivo Targeting and Imaging of Atherosclerosis Using Multifunctional Virus-Like Particles of Simian Virus 40. Nano Lett 2016, 16, 6164-6171. [CrossRef]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, Indicators, Risk Factors and New Hopes. Int J Prev Med 2014, 5, 927-946.
- Mulder, W.J.; Jaffer, F.A.; Fayad, Z.A.; Nahrendorf, M. Imaging and Nanomedicine in Inflammatory Atherosclerosis. Sci Transl Med 2014, 6, 239sr231. [CrossRef]
- Cassette, E.; Helle, M.; Bezdetnaya, L.; Marchal, F.; Dubertret, B.; Pons, T. Design of New Quantum Dot Materials for Deep Tissue Infrared Imaging. Adv Drug Deliv Rev 2013, 65, 719-731. [CrossRef]
- Aanei, I.L.; ElSohly, A.M.; Farkas, M.E.; Netirojjanakul, C.; Regan, M.; Taylor Murphy, S.; O'Neil, J.P.; Seo, Y.; Francis, M.B. Biodistribution of Antibody-MS2 Viral Capsid Conjugates in Breast Cancer Models. Mol Pharm 2016, 13, 3764-3772. [CrossRef]
- ElSohly, A.M.; Netirojjanakul, C.; Aanei, I.L.; Jager, A.; Bendall, S.C.; Farkas, M.E.; Nolan, G.P.; Francis, M.B. Synthetically Modified Viral Capsids as Versatile Carriers for Use in Antibody-Based Cell Targeting. Bioconjug Chem 2015, 26, 1590-1596. [CrossRef]
- Hu, H.; Masarapu, H.; Gu, Y.; Zhang, Y.; Yu, X.; Steinmetz, N.F. Physalis Mottle Virus-like Nanoparticles for Targeted Cancer Imaging. ACS Appl Mater Interfaces 2019, 11, 18213-18223. [CrossRef]
- Naresh, V.; Lee, N. A Review on Biosensors and Recent Development of Nanostructured Materials-Enabled Biosensors. Sensors (Basel) 2021, 21. [CrossRef]
- Mieloch, A.A.; Mleczko, A.M.; Samelak-Czajka, A.; Jackowiak, P.; Rybka, J.D. Biomimetic Virus-Like Particles with Magnetic Core. From Bioactivity to an Immunodiagnostic Tool. Chemical Engineering Journal 2024, 149714. [CrossRef]
- Zhang, Q.; Su, C.; Qiu, J.-G.; Jiang, B.-H.; Zhang, C.-y. Advances in Enzyme-Free Nucleic Acid Amplification-Based Fluorescent Biosensors for Real-Time Imaging of DNA Repair Enzymes in Living Cells. Coordination Chemistry Reviews 2023, 496, 215406.
- Ye, M.; Hu, S.; Zhou, L.; Tang, X.; Zhao, S.; Zhao, J. Fluidic Membrane Accelerating the Kinetics of Photoactivatable Hybridization Chain Reaction for Accurate Imaging of Tumor-Derived Exosomes. Anal Chem 2022, 94, 17645-17652. [CrossRef]
- Dirks, R.M.; Pierce, N.A. Triggered Amplification by Hybridization Chain Reaction. Proceedings of the National Academy of Sciences 2004, 101, 15275-15278. [CrossRef]
- Chen, Y.; Song, Y.; Wang, X.; Tang, H.; Li, C. Genetically Engineered Virus-Like Particle-Armoured and Multibranched DNA Scaffold-Corbelled Ultra-Sensitive Hierarchical Hybridization Chain Reaction for Targeting-Enhanced Imaging in Living Biosystems Under Spatiotemporal Light Powering. Biosens Bioelectron 2024, 247, 115943. [CrossRef]
- Wang, J.; Zhang, L.; Yan, G.; Cheng, L.; Zhang, F.; Wu, J.; Lei, Y.; An, Q.; Qi, H.; Zhang, C.; et al. Modified Exfoliated Graphene Functionalized with Carboxylic Acid-Group and Thionine on a Screen-Printed Carbon Electrode as a Platform for an Electrochemical Enzyme Immunosensor. Mikrochim Acta 2024, 191, 143. [CrossRef]
- Lua, L.H.; Connors, N.K.; Sainsbury, F.; Chuan, Y.P.; Wibowo, N.; Middelberg, A.P. Bioengineering Virus-Like Particles as Vaccines. Biotechnol Bioeng 2014, 111, 425-440. [CrossRef]
- Hillebrandt, N.; Hubbuch, J. Size-Selective Downstream Processing of Virus Particles and Non-Enveloped Virus-Like Particles. Front Bioeng Biotechnol 2023, 11, 1192050. [CrossRef]
- Roldao, A.; Mellado, M.C.; Lima, J.C.; Carrondo, M.J.; Alves, P.M.; Oliveira, R. On the Effect of Thermodynamic Equilibrium on the Assembly Efficiency of Complex Multi-Layered Virus-Like Particles (VLP): the Case of Rotavirus VLP. PLoS Comput Biol 2012, 8, e1002367. [CrossRef]
- Zhao, Q.; Allen, M.J.; Wang, Y.; Wang, B.; Wang, N.; Shi, L.; Sitrin, R.D. Disassembly and Reassembly Improves Morphology and Thermal Stability of Human Papillomavirus Type 16 Virus-Like Particles. Nanomedicine 2012, 8, 1182-1189. [CrossRef]
- Zhao, Q.; Modis, Y.; High, K.; Towne, V.; Meng, Y.; Wang, Y.; Alexandroff, J.; Brown, M.; Carragher, B.; Potter, C.S.; et al. Disassembly and Reassembly of Human Papillomavirus Virus-Like Particles Produces More Virion-Like Antibody Reactivity. Virol J 2012, 9, 52. [CrossRef]
- Gupta, R.; Arora, K.; Roy, S.S.; Joseph, A.; Rastogi, R.; Arora, N.M.; Kundu, P.K. Platforms, Advances, and Technical Challenges in Virus-Like Particles-Based Vaccines. Front Immunol 2023, 14, 1123805. [CrossRef]
- Tariq, H.; Batool, S.; Asif, S.; Ali, M.; Abbasi, B.H. Virus-Like Particles: Revolutionary Platforms for Developing Vaccines Against Emerging Infectious Diseases. Front Microbiol 2021, 12, 790121. [CrossRef]
- Fuenmayor, J.; Godia, F.; Cervera, L. Production of Virus-Like Particles for Vaccines. N Biotechnol 2017, 39, 174-180. [CrossRef]
- Fernandes, B.; Correia, R.; Alves, P.M.; Roldao, A. Intensifying Continuous Production of Gag-HA VLPs at High Cell Density Using Stable Insect Cells Adapted to Low Culture Temperature. Front Bioeng Biotechnol 2022, 10, 917746. [CrossRef]
- Fontana, D.; Kratje, R.; Etcheverrigaray, M.; Prieto, C. Immunogenic Virus-Like Particles Continuously Expressed in Mammalian Cells as a Veterinary Rabies Vaccine Candidate. Vaccine 2015, 33, 4238-4246. [CrossRef]
- Comas-Garcia, M.; Kroupa, T.; Datta, S.A.; Harvin, D.P.; Hu, W.S.; Rein, A. Efficient Support of Virus-Like Particle Assembly by the HIV-1 Packaging Signal. Elife 2018, 7. [CrossRef]
- Mohsen, M.O.; Gomes, A.C.; Vogel, M.; Bachmann, M.F. Interaction of Viral Capsid-Derived Virus-Like Particles (VLPs) with the Innate Immune System. Vaccines (Basel) 2018, 6. [CrossRef]
- Tornesello, A.L.; Tagliamonte, M.; Buonaguro, F.M.; Tornesello, M.L.; Buonaguro, L. Virus-Like Particles as Preventive and Therapeutic Cancer Vaccines. Vaccines (Basel) 2022, 10. [CrossRef]
- He, J.; Yu, L.; Lin, X.; Liu, X.; Zhang, Y.; Yang, F.; Deng, W. Virus-Like Particles as Nanocarriers for Intracellular Delivery of Biomolecules and Compounds. Viruses 2022, 14. [CrossRef]
- Woolf, T.M. Therapeutic Repair of Mutated Nucleic Acid Sequences. Nat Biotechnol 1998, 16, 341-344. [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-Frequency Off-Target Mutagenesis Induced by CRISPR-Cas Nucleases in Human Cells. Nat Biotechnol 2013, 31, 822-826. [CrossRef]
- Gaj, T.; Sirk, S.J.; Shui, S.L.; Liu, J. Genome-Editing Technologies: Principles and Applications. Cold Spring Harb Perspect Biol 2016, 8. [CrossRef]
- Lindel, F.; Dodt, C.R.; Weidner, N.; Noll, M.; Bergemann, F.; Behrendt, R.; Fischer, S.; Dietrich, J.; Cartellieri, M.; Hamann, M.V.; et al. TraFo-CRISPR: Enhanced Genome Engineering by Transient Foamy Virus Vector-Mediated Delivery of CRISPR/Cas9 Components. Mol Ther Nucleic Acids 2019, 18, 708-726. [CrossRef]
- Lyu, P.; Javidi-Parsijani, P.; Atala, A.; Lu, B. Delivering Cas9/sgRNA Ribonucleoprotein (RNP) by Lentiviral Capsid-Based Bionanoparticles for Efficient ‘Hit-and-Run' Genome Editing. Nucleic Acids Res 2019, 47, e99. [CrossRef]
- Choi, J.G.; Dang, Y.; Abraham, S.; Ma, H.; Zhang, J.; Guo, H.; Cai, Y.; Mikkelsen, J.G.; Wu, H.; Shankar, P.; et al. Lentivirus Pre-Packed with Cas9 Protein for Safer Gene Editing. Gene Ther 2016, 23, 627-633. [CrossRef]
- Knopp, Y.; Geis, F.K.; Heckl, D.; Horn, S.; Neumann, T.; Kuehle, J.; Meyer, J.; Fehse, B.; Baum, C.; Morgan, M.; et al. Transient Retrovirus-Based CRISPR/Cas9 All-in-One Particles for Efficient, Targeted Gene Knockout. Mol Ther Nucleic Acids 2018, 13, 256-274. [CrossRef]
- Cai, Y.; Bak, R.O.; Mikkelsen, J.G. Targeted Genome Editing by Lentiviral Protein Transduction of Zinc-Finger and TAL-Effector Nucleases. Elife 2014, 3, e01911. [CrossRef]
- Habeeb, M.; You, H.W.; Umapathi, M.; Ravikumar, K.K.; Hariyadi; Mishra, S. Strategies of Artificial intelligence Tools in the Domain of Nanomedicine. Journal of Drug Delivery Science and Technology 2024, 91. [CrossRef]
- Li, R.; Wijma, H.J.; Song, L.; Cui, Y.; Otzen, M.; Tian, Y.; Du, J.; Li, T.; Niu, D.; Chen, Y.; et al. Computational Redesign of Enzymes for Regio- and Enantioselective Hydroamination. Nat Chem Biol 2018, 14, 664-670. [CrossRef]
- Cunningham, J.M.; Koytiger, G.; Sorger, P.K.; AlQuraishi, M. Biophysical Prediction of Protein-Peptide Pnteractions and Signaling Networks Using Machine Learning. Nat Methods 2020, 17, 175-183. [CrossRef]
- Madhukar, N.S.; Khade, P.K.; Huang, L.; Gayvert, K.; Galletti, G.; Stogniew, M.; Allen, J.E.; Giannakakou, P.; Elemento, O. A Bayesian Machine Learning Approach for Drug Target Identification Using Diverse Data Types. Nat Commun 2019, 10, 5221. [CrossRef]
- Ianevski, A.; Giri, A.K.; Gautam, P.; Kononov, A.; Potdar, S.; Saarela, J.; Wennerberg, K.; Aittokallio, T. Prediction of Drug Vombination Effects with a Minimal Set of Experiments. Nat Mach Intell 2019, 1, 568-577. [CrossRef]
- Gainza, P.; Sverrisson, F.; Monti, F.; Rodola, E.; Boscaini, D.; Bronstein, M.M.; Correia, B.E. Deciphering Interaction Fingerprints from Protein Molecular Surfaces Using Geometric Deep Learning. Nat Methods 2020, 17, 184-192. [CrossRef]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in Cancer Therapy: Challenges, Opportunities, and Clinical Applications. J Control Release 2015, 200, 138-157. [CrossRef]
- Padma, V.V. An Overview of Targeted Cancer Therapy. Biomedicine (Taipei) 2015, 5, 19. [CrossRef]
- Verhoef, J.J.; Anchordoquy, T.J. Questioning the Use of PEGylation for Drug Delivery. Drug Deliv Transl Res 2013, 3, 499-503. [CrossRef]
- Gulati, N.M.; Stewart, P.L.; Steinmetz, N.F. Bioinspired Shielding Strategies for Nanoparticle Drug Delivery Applications. Mol Pharm 2018, 15, 2900-2909. [CrossRef]
Figure 1.
Relevant features of VLPs for biomedical applications [23].
Figure 1.
Relevant features of VLPs for biomedical applications [23].
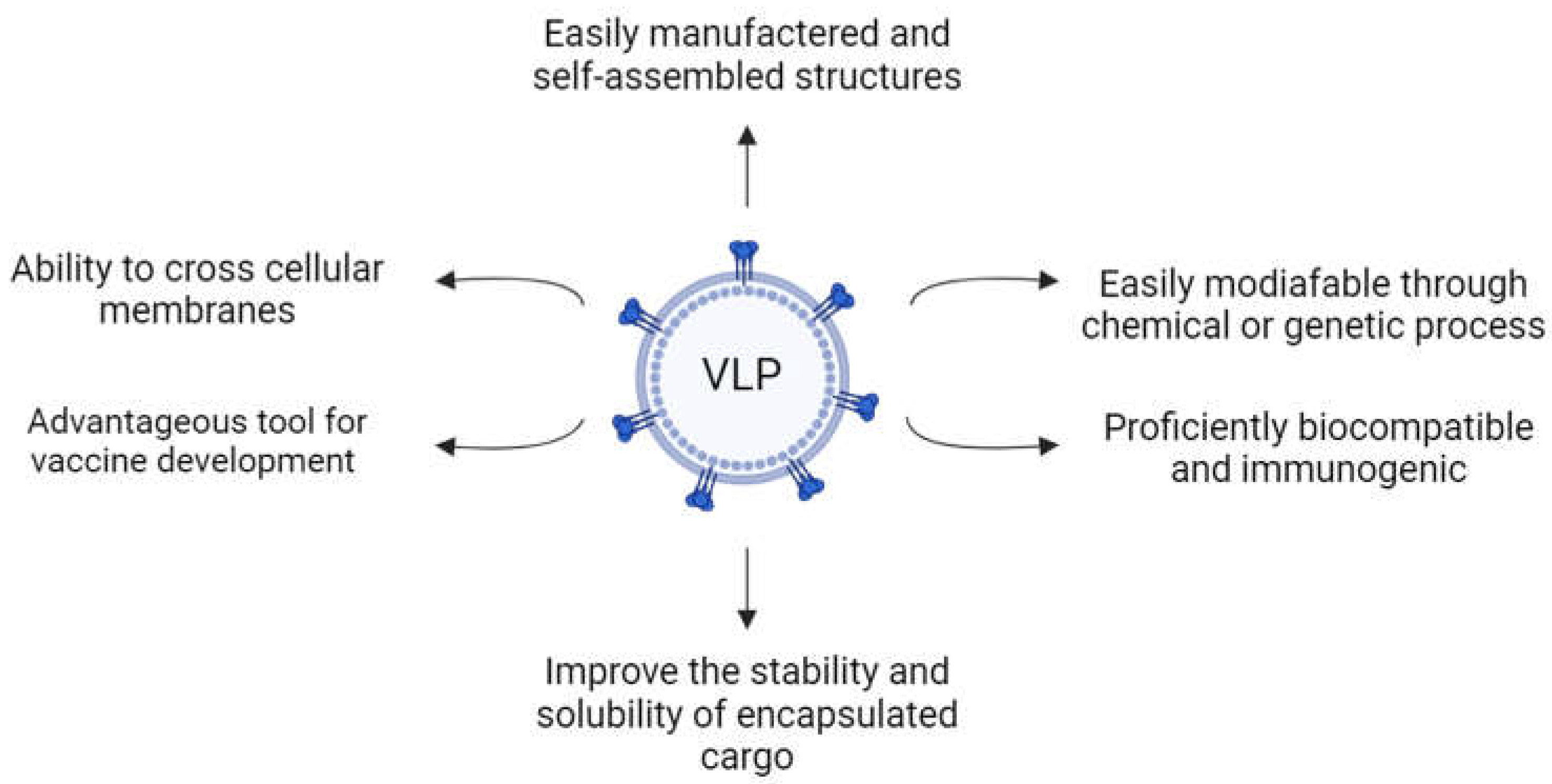

Figure 3.
Downstream process approach for VLP purification [64].
Figure 3.
Downstream process approach for VLP purification [64].
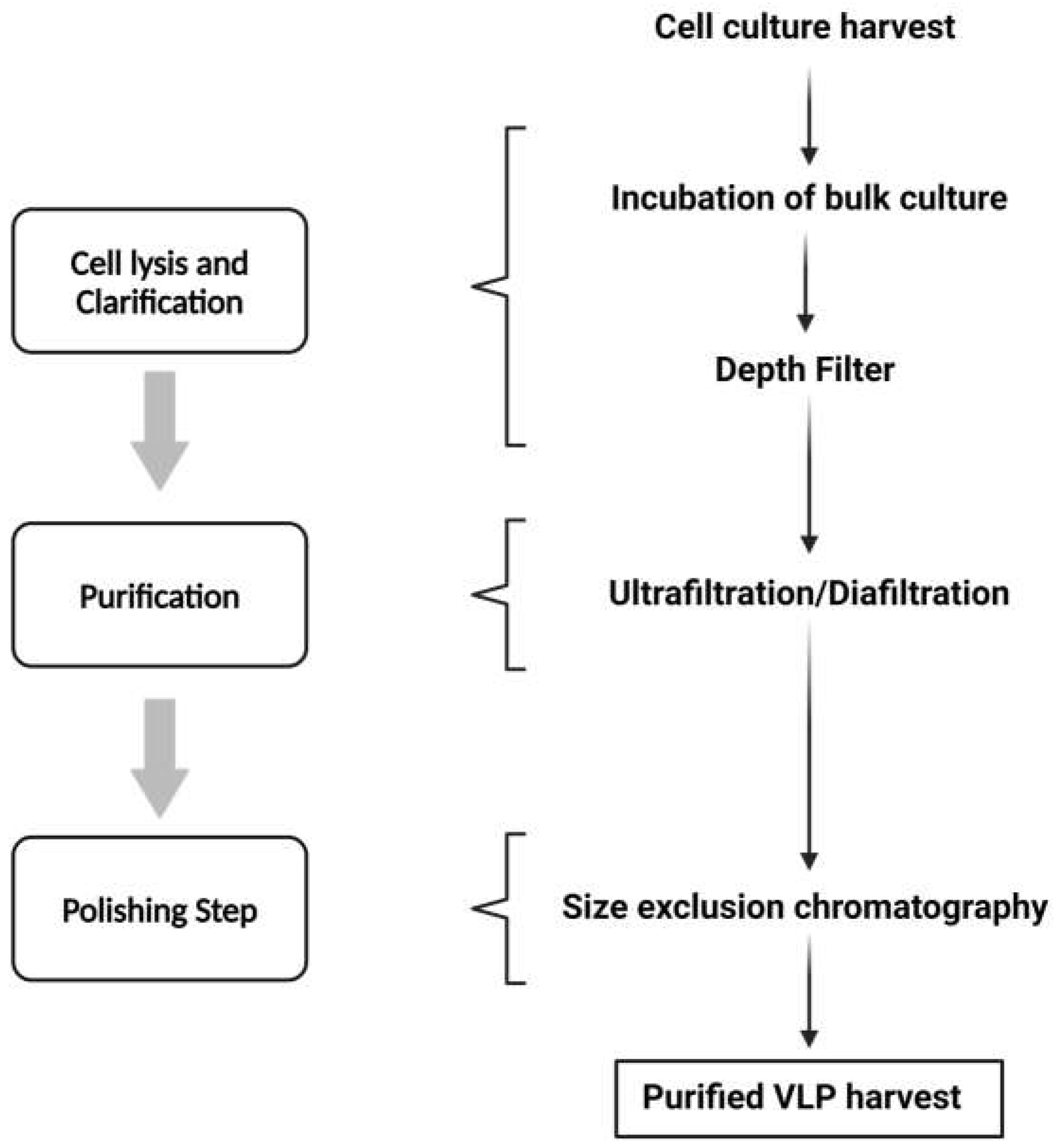
Figure 4.
Methods for functionalizing the surface of VLPs.


Table 1.
Representative table of different VLPs, their characteristics and applications.
| Class | VLP | Virus Type | Expression Systems | Applications | References |
|---|---|---|---|---|---|
| Enveloped | HIV | Human virus | Eukaryotic (Mammalian, Yeast, Insect, Plant) | Vaccination and Drug Delivery | [25] |
| Influenza | Human virus | Eukaryotic (Mammalian, Insect, Plant) | Vaccination | [45] | |
| Ebola | Human virus | Eukaryotic (Mammalian, Insect) | Vaccination | [46] | |
| Dengue | Human virus | Eukaryotic (Mammalian, Yeast, Insect, Plant) | Vaccination and Diagnosis | [47] | |
| Non-Enveloped | HPV | Human virus | Prokaryotic and Eukaryotic (Mammalian, Yeast, Insect, Plant) | Vaccination and Drug Delivery | [24] |
| Norwalk | Human virus | Prokaryotic and Eukaryotic (Mammalian, Insect, Plant) | Vaccination | [26] | |
| Hepatitis B (core) | Human virus | Prokaryotic and Eukaryotic (Mammalian, Insect, Plant) | Vaccination, Drug Delivery | [48] | |
| MS2 | Bacteriophage | Prokaryotic and Eukaryotic (Yeast, | Vaccination, Drug Delivery and Imaging | [49] | |
| P22 | Bacteriophage | Prokaryotic | Vaccination, Drug Delivery and Imaging | [50] | |
| Qβ | Bacteriophage | Prokaryotic and Eukaryotic (Yeast) | Vaccination, Drug Delivery and Imaging | [51] |
Table 2.
Representative table of Food and Drug Administration-approved virus-like particles.
| Virus | Recombinant Protein | Expression System | VLP Type | Vaccine | Reference |
|---|---|---|---|---|---|
| Hepatitis B virus (HBV) | HBsAg | Eukaryotic Cell (S. cerevisiae) | Non-Enveloped | Engerix-B® | [116] |
| Eukaryotic Cell (P. pastoris) | Non-Enveloped | Enivac HB® | [117] | ||
| Eukaryotic Cell (S. cerevisiae) | Non-Enveloped | Euvax® | [118] | ||
| Eukaryotic Cell (H. polymorpha) | Non-Enveloped | Gene Vac-B® | [119] | ||
| Eukaryotic Cell (P. pastoris) | Non-Enveloped | Heberiovac HB | [120] | ||
| Eukaryotic Cell (H. polymorpha) | Non-Enveloped | Hepavax-Gene® | [121] | ||
| Eukaryotic Cell (S. cerevisiae) | Non-Enveloped | Recombivax HB® | [122] | ||
| Eukaryotic Cell (P. pastoris) | Non- Enveloped | Revac-B® | [123] | ||
| Eukaryotic Cell (P. pastoris) | Non-Enveloped | Shanvac-B® | [124] | ||
| Papillomavirus | HPV6 /11/16/18 L1 | Eukaryotic Cell (S. cerevisiae) | Non-Enveloped | Gardasil® | [125] |
| HPV6 16/18 L1 | Insect Cells | Non-Enveloped | Cervarix® | [126] | |
| Influenza virus A | A (H1N1), A (H3N2), B, HA, NA |
Cell-Free | Enveloped | Inflexal® V | [127] |
Table 3.
Different expression systems for VLP production and their advantages and disadvantages [153,155].
| Expression System | Advantages | Disadvantages |
|---|---|---|
| Bacteria |
|
|
| Yeast |
|
|
| Insect cells |
|
|
| Mammalian cells |
|
|
| Plant cells |
|
|
| Cell-free systems |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.


Nanoparticle-Based Secretory Granules Induce a Specific and Long-Lasting Immune Response through Prolonged Antigen Release
Laia Bosch-Camós
et al.
,
2024
Modulation of Antigen Display on PapMV Nanoparticles Influences Its Immunogenicity
Marie-Eve Laliberté-Gagné
et al.
,
2020
The Adjuvant Activity of BCG Cell Wall Cytoskeleton on Dengue Virus-2 Subunit Vaccine
Tuksin Jearanaiwitayakul
et al.
,
2023



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated