
Preprint
Article
Selection and Multiplexing of Reverse Transcription Quantitative PCR Tests Targeting Relevant Honeybee Viral Pathogens
Altmetrics
Downloads
97
Views
55
Comments
0
A peer-reviewed article of this preprint also exists.



This version is not peer-reviewed
Submitted:
03 May 2024
Posted:
06 May 2024
You are already at the latest version
Alerts
Abstract
Verification of inclusivity of molecular detection methods gives indications on the reliability of viral infections diagnosis because of the tendency of viral pathogens to undergo sequence variation. This study was aimed to select inclusive probe based reverse transcription quantitative PCR (RT-qPCR) assays for the diagnosis of the most widespread and detrimental viruses infecting honeybees, namely the acute bee paralysis virus (ABPV), the black queen cell virus (BQCV), the chronic paralysis bee virus (CBPV), the deformed wing virus variants A (DWVA) and B (DWVB), and the sacbrood virus (SBV). Therefore, detection methods previously described were re-evaluated in silico for specificity and inclusivity. Based on this evaluation, selected methods were modified or new ones were designed and tested in duplex RT-qPCR reactions. The limits of detection (LODs), effect of multiplexing on sensitivity and viral RNA quantification potential in bees and hive debris were assessed. This study made available diagnostic assays able to detect an increased number of virus variants compared to previously described tests and two viral pathogens in a single PCR reaction.
Keywords:
Subject: Biology and Life Sciences - Life Sciences
1. Introduction
Honeybee health is threatened worldwide by different detrimental factors that determine annual mortality rates ranging between 9.6 and 26% for domesticated honeybees Apis mellifera and A. cerana at extents varying with the geographical area [1]. Different viral pathogens contribute to honeybee mortality by causing acute infections or by acting as biotic stressors [2,3]. In particular, colony losses for A. mellifera, the most important commercial pollinator, occur mainly overwinter and have been strongly associated to the deformed wing virus (DWV) transmission by V. destructor, since this allows direct access of the virus in bee haemolymph causing infections very rapidly. It is widely accepted that DWV is a good indicator of bee colony decline for its positive temporal correlation with honeybee colony losses [4]. In addition, the DWV induces immune suppression in bees facilitating the ectoparasitic trophic activity of the Varroa destructor mite [3].
Viruses affecting honeybees are able to replicate in different organs with broad or restricted tropism and impair organ function when they are present at very high titers giving rise to infection symptoms. Asymptomatic infections remain undetected and may cause long-term damage to the honeybee colonies [5].
The most often detected viruses in honeybees are the DWV in the two variants A (DWVA) and B (DWVB), the latter originally named Varroa destructor virus-1 (VDV-1) [2,6], the chronic bee paralysis virus (CBPV), the black queen cell virus (BQCV), the sacbrood virus (SBV), and the acute bee paralysis virus (ABPV) [2,7,8,9,10,11,12,13,14,15,16]. ABPV is the most common of the viruses causing acute paralysis, i.e., ABPV, Israeli acute paralysis virus (IAPV) and Kashmir bee virus (KBV) in Europe and South America and was detected globally, with exception of Australia [13,17,18,19]. IAPV was suspected but not confirmed to be responsible for the colony collapse disorder (CCD), characterized by a sudden loss of up to 90% hives in apiaries without a clear precedent history of disease. However, the involvement of the closely related ABPV and KBV cannot be excluded for the possibility of misidentification [20].
Among these most common viruses, DWVB and CBPV showed increasing prevalence in many geographical areas [9,21,22,23,24,25]. In particular, the relative representation of DWVB in thousand RNAseq libraries for A. mellifera, Bombus terrestris and V. destructor increased after 2014 in US and became dominant in Europe [24].
The viruses DWVA, DWVB, BQCV, CBPV, SBV and ABPV are positive-sense single-stranded (+ss) RNA viruses of the families Dicistroviridae and Iflaviridae, except for CBPV, which remains unassigned [14,20, https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=180822, accessed on 2 April 2024]. These threaten beekeeping worldwide and give rise to a multiplicity of symptoms [20]. In particular, DWV infected bees show crumpled or aborted wings, shortened abdomens, paralysis, severely shortened life span for workers and drones, impaired learning and foraging behavior. SBV causes pupation failure, the formation of swollen larvae (’sac’) filled with ecdysial fluid containing numerous viral particles, impaired foraging, reduction of adult life span and metabolic activities. BQCV affects queen larvae that become initially yellowish with sac-appearance and then dark brown, causes the death of infected pupae and the significant reduction of the adult bee life span. CBPV infected bees appear trembling, with bloated abdomen, unable to fly and crawling on the ground. Alternatively, bees appear hairless, darker and shiny, are attacked by the healthy bees. In both cases the infected bees die in a few days. ABPV infection manifests with trembling bees unable to fly, darkened and hairless, crawling on the ground before rapid death. ABPV, DWVA and DWVB are transmitted by V. destructor [2]. Moreover, it was reported that mite presence was significantly associated with higher prevalence of BQCV, CBPV, and SBV [26].
McMahon et al. [4] reported that DWVB is more virulent than DWVA in adult worker bees exposed to field-derived DWVA and DWVB in the absence of V. destructor and cause a faster colony collapse. On the contrary, Norton et al. [27] found evidence that the DWVB variant caused a lower mortality in honeybee pupae than DWVA and it accumulated to higher levels. This might explain the increasing DWVB prevalence over DWVA reported in different countries [21,22,23,24,25]. Moreover, Kevill et al. [23] reported that DWVA is transmitted by V. destructor, while DWVB is not. In addition, the latter caused less severe infections than DWVA and might be able to protect bee colonies from the DWVA infection by superinfection exclusion [23]. In addition, DWVB appeared to be detrimental for V. destructor since mites with high DWVB loads had an average lifespan significantly shorter than those with high DWVA levels or with low levels of DWVA or DWVB. This could be a consequence of the different ability of DWVB and DWVA to replicate in V. destructor, though there is not general agreement on this aspect [28,29,30].
Based on the reported differences in pathogenicity between DWVA and DWVB variants, use of molecular detection methods able to differentiate between the two variants is preferable in studies regarding virulence and effects on bee colony health. However, many of the DWV targeted RT-qPCR tests reported either do not discriminate between the two variants or target only the DWVA variant [11,15,31,32,33].
The RT-qPCR based methods available for the detection of ABPV, BQCV, CBPV, DWVA, DWVB and SBV are singleplex tests that use Sybr Green [25,26,29,31,32,34,35,36,37,38] or fluorescent probe based detection [33,39,40,41,42] but only two were validated [6,40].
Most tests were already applied in ecological studies and diagnostic screenings [4,9,25,43,44,45,46,47,48].
Limitedly to the DWV virus, the analysis of published genome sequences suggested that PCR/qPCR detection of the virus can be unreliable since variant detection depends on the choice of appropriate oligonucleotide primers [24]. Therefore, this study was aimed to investigate in silico the ability of the described RT-qPCR tests for viruses ABPV, BQCV, CBPV, DWVA, DWVB and SBV to be specific and inclusive. The ones theoretically more reliable were selected to derive new methods with improved inclusivity. For some of the viral pathogens new methods were designed.
2. Materials and Methods
2.1. Oligonucleotide Selection/Design
The primers and probe systems previously described for the detection of honeybee viral pathogens ABPV, BQCV, DWVA, DWVB, CPBV and SBV [25,26,29,31,32,33,34,35,36,37,38,39] were tested for specificity and inclusivity by BLASTN (https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 27 April 2024) against up to 500 database entries. Those matching the highest number of sequences of the target virus were selected for use in one step RT-qPCR tests with TaqMan probes. When opportune, the primers were modified to increase the number of matched sequences and improve inclusivity. TaqMan probes were designed for tests originally conceived for Sybr Green detection. Primers and probes used in this study were synthetized by Eurofins Genomics (Ebersberg, Germany). Their tendency to dimerize and optimal use in qPCR tests were analyzed by the online tools available from Eurofins Genomics (https://eurofinsgenomics.eu/en/dna-rna-oligonucleotides/oligo-tools, accessed on 23 December 2023).
2.2. RNA Extraction
RNA was extracted from two honeybees or an approximate volume 800 µl of hive debris. These were transferred in 2 ml Eppendorf safe-lock tubes (Eppendorf, Milan, Italy) containing approximately a volume of 200 µl of unwashed glass beads with 200 µm diameter (Merck) sterilized by autoclaving. The hive debris were added until reaching the 1 ml graduation of the tube. Hive debris weighing was not carried out since its practical significance is limited for the extremely high variability in composition and density.
One ml of Macherey Nagel Nucleozol reagent (Carlo Erba, Cornaredo, MI, Italy) was added. Bees were chopped with a serological pipette, then the suspensions of bees and hive debris were bead-beaten in a TissueLyser II (Qiagen) at 30 hz for 2 min.
The homogenate was centrifuged at 14,000 x g for 10 min at 4°C and the supernatant was transferred in an Eppendorf tube containing 1 ml of ice-cold isopropanol (Merck), 25 µg of RNA extraction control homogenized in RNAse-free water and 5 µg of Poly-A carrier RNA (Qiagen, Milan, Italy). The RNA extraction control was constituted by Tenebrio molitor larvae obtained from a lyophilized food preparation intended for human consumption (Tarme della Farina Liofilizzate, ZIRP Insects, Vienna, Austria) and previously tested for the absence of amplification with the honeybee virus targeted RT-qPCR assays. Aliquots of the RNA extraction control preparation were stored at -80°C for future usage. The mixture was agitated by inverting the tube and kept on ice for 30 min. Then it was centrifuged at 14,000 x g for 10 min and the supernatant was discarded. The pellet was resuspended in 500 µl of Macherey Nagel MDB buffer (Carlo Erba) and loaded into a New England Biolabs Monarch RNA Cleanup Column (Euroclone, Pero, MI, Italy). The column was centrifuged for 1 min at 11,000 x g and the eluate was discarded, then the column was washed with 600 µl of Macherey Nagel RAW2 buffer (Carlo Erba) and with 600 µl of ice cold 75% ethanol prepared with RNAse-free water. The ethanol was completely removed by centrifugation at 11,000 x g for 2 min and the RNA was eluted in two steps with 30 µl of RNAse-free water. The RNA was soon analyzed by RT-qPCR or stored at -80°C until analysis.
2.3. RT-qPCR
RT-qPCR was carried out in a QuantStudio 5 thermal cycler (Thermo Fisher Scientific) by using the One Step PrimeScript III RT-PCR Kit Takara Bio (Diatech, Jesi, AN, Italy) master mix. The 20 µl reaction contained 10 µl of master mix, 0.05 µg/µl BSA, 1× ROX, 0.2 µM of each primer and probe for the viral targets, and 0.1 µM of each primer and probe for the RNA extraction control derived from those described by Köppel et al. [49] TeneF: 5′-CCATGAGTACGAATAAGAGAAACCAA-3′, TeneR: 5′-TTTAAGGCTTGAATTTGTTGTTTTATCTGTTTATT-3′ and the TeneP: 5′-JOE-AATAGATAGACCAAGAACGCCTTCACA-BHQ13′ probe, 4 µl of RNA extract and RNAse-free water to adjust the reaction volume. A negative extraction control obtained without the sample and a positive control reaction containing 6 Log copies of synthetic RNA were included in each PCR run.
The PCR program was unique for all the targets and comprised 15 min at 50°C for reverse transcription, 2 min of denaturation at 95°C and 50 cycles of denaturation at 95°C for 15 sec and annealing at 54°C for 30 sec.
2.4. Determination of the Limit of Detection and Construction of Calibration Curves
The limit of detection (LOD) of the RT-qPCR tests was determined by using RNA fragments synthetized by GenScript Biotech (Rijswijk, The Netherlands) with sequences identical to the viral target region. The synthetic RNA fragments were quantified by the Qubit 3 Fluorometer (Thermo Fisher Scientific, Rodano, MI, Italy) with the Qubit RNA HS Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions and used to spike honeybee or hive debris samples which in preliminary assays tested negative in the RT-qPCR tests to be evaluated in this study. The copy number of synthetic RNA was determined based on the concentration and length by using the online tool https://nebiocalculator.neb.com/#!/ssrnaamt (accessed on 25 January 2024). Serial dilutions of the synthetic RNA fragments were used to construct calibration curves by spiking honeybee or hive debris aliquots in triplicate.
2.5. Statistical Analyses
The Student’s t test on Ct values was used to evaluate the effect of multiplexing on amplification yield and sensitivity using Microsoft Excel 2016.
3. Results
3.1. In Silico Assessment of Existing Honeybee Virus Detection Methods
The outcomes of the in silico evaluation for specificity and inclusivity of previously described methods suggested to design new tests for ABPV and CBPV, and to modify of existing methods for BQCV, DWVA, DWVB and SBV in order to include an higher number of virus variants.
For BQCV the primers and probe were derived from those described by Chantawannakul et al. [39] and were shortened to fit the annealing temperature of the unique amplification program used for all the assays evaluated in this study. Primers and probes targeting DWVA and DWVB were in part derived from those described by Kevill et al. [38]. In particular, the same specific reverse primers were used, while the forward primers were re-designed to be specific for each DWV variant. Moreover, probes intended to be specific for each variant were newly designed, since the original test was based on Sybr Green detection. The primers and probes for SBV detection were derived from those validated by Blanchard et al. [41] but the forward primer was shortened of one nucleotide at the 3′ end and a degenerate position was introduced at the third nucleotide position to increase the number of matched GenBank entries. Moreover, the reverse primer was newly designed and the probe was extended by adding ten nucleotides at the 3′ end. All other probes used in this study were of the Minor Groove Binding (MGB) type. The inclusivity of the CBPV targeted probe was tested by BLAST runs with all the possible versions without degenerate nucleotide positions.
The primer/probes systems evaluated in this study are reported in Table 1.
The number of database entries perfectly matched by the primers reported in Table 1 were 91 for the ABPV specific test, 496 for the BQCV specific test, 110 for the CBPV specific test, 256 for the DWVA specific test, 129 for the DWVB specific test and 232 for the SBV specific test. These matched entry numbers correspond to those of the primer with the lowest number of database matches in each test. For all the specific tests the theoretical number of matched sequences was higher than for the previously reported assays. The DWVA specific test, like those reported previously, also matched the Kakugo variant [50].
3.2. Multiplexing of RT-qPCR Tests Targeting Honeybee Viruses
The RT-qPCR assays designed in this study were tested for multiplexing potential in order to reduce the number of analyses necessary to diagnose the six viral pathogens considered. Therefore, duplex reactions were tested on synthetic target RNAs combined in the same concentration or with one in large excess compared to the other.
The RNA extraction control used in the reactions was constituted a homogenate of T. molitor. Use of this organism was decided based on the possibility to extend its future use also to honeybee insect parasites and on the low probability of its presence in hives. Duplex RT-qPCR reactions run without the synthetic RNA targets demonstrated the absence of false positive results deriving from the RNA extraction control. This insect was purchased in form of commercial food preparation for the unavailability of collections providing it.
The best performing RT-qPCR assays combinations were ABPV with BQCV, DWVA with DWVB, and CBPV with SBV. The Student’s t test indicated that when the templates were present in the same concentration the difference between series of Ct values obtained in triplicate reactions containing only one or both targets in most cases was not statistically significant over the range 1 – 6 Log copy number. The P values obtained for the Ct comparison of each target alone or in association with the other target at the different concentrations are reported in Table 2.
As shown, statistically different Ct series for P<0.05 were obtained only for the DWVA targeted test at the lowest concentration. This was caused by the increase of Ct values in the duplex reaction. However, also in the duplex reaction the target virus was detected in all replicates at the lowest concentration, so sensitivity did not vary compared to the reaction with the DWVA target alone. The experiments of amplification yield comparison between reactions containing one target or two were carried out using both the primer/probe systems for the duplex test also when only one target was present. This allowed to highlight that the DWVA and DWVB targeted tests were not specific for the intended variant. Indeed, the DWVA targeted test gave rise to an apparent DWVB specific amplification at Ct values 8±1 lower than those of the DWVA target when only the latter was present. Moreover, the DWVB specific test gave rise to an apparent DWVA specific amplification at Ct values 4±1 lower than those of the DWVB target. This could be a consequence of the lack of specificity of the probes when the amplification product accumulates, despite it was not evident in the in silico assessment of specificity. Duplex RT-qPCR runs carried out with one of the two targets in higher concentration allowed to determine that DWVB cannot be specifically detected in real samples when present at more than 2 Log copy number lower than DWVA, and DWVA cannot be determined in real samples by the duplex reaction when present at more than 1 Log copy number lower than DWVA. Nonetheless, the duplex test identifies the DWV variant that predominates in a sample and can detect coinfection with DWVA and DWVB when the latter is not lower than 2 Log copy number compared to DWVA and when DWVA is not lower than 1 Log copy number compared to DWVB. The detection of both variants was not affected when these were present at the same concentration, thus allowing the construction of calibration curves. For the reliable detection and quantification of the two DWV variants in real samples use of separate singleplex reactions is advisable.
It was also observed that the BQCV specific test presented abnormal amplifications curves for template amounts higher than 6 Log copy number. Indeed, the amplification curve showed a normal trend until the beginning of the plateau phase and then dropped with apparently negative final results. This was observed, though less frequently, also when the Cy5 labelled probe was replaced with a FAM labelled probe. Therefore, when using the BQCV specific test it is opportune to register the Ct values for the BQCV manually while running at the beginning of the exponential phase in the analysis of real samples.
3.3. Limit of Detection (LOD) of the RT-qPCR Tests
The LOD of the RT-qPCR tests designed in this study was determined both in terms of template copy number detectable in the PCR reaction and in terms of Log copy number spiked in bee and hive debris samples. According to the minimum requirement guidelines (MIQE) for the publication of qPCR tests [Bustin], the LOD95 was defined. Therefore, 20 replicate reactions using synthetic RNA targets or 20 extractions from spiked bees and hive debris were carried out followed by the RT-qPCR tests to define the LOD. To this aim five extraction/amplification replicates were preliminarily run for decreasing target concentrations and the lowest concentration allowing amplification from all five extracts was finally used in extractions/amplifications from 20 replicates. LOD values obtained for each viral target in the duplex reactions are shown in Table 3.
The LOD was confirmed also in duplex reactions with different concentrations of the two templates to define if competition between targets could decrease sensitivity. In these duplex reactions one template was present in 5 Log copy number more than the target at LOD concentration and no loss in sensitivity was observed for the latter.
3.4. Virus Quantification in Artificially Spiked Samples
Calibration curves for the quantification of viral loads by the duplex RT-qPCR reactions targeted on ABPV/BQCV, DWVA/DWVB and CBPV/SBV were constructed for honeybees and hive debris and presented a linearity range encompassing at least five decimal dilutions of the target synthetic RNA. Examples are shown in Figure 1.
Results showed that the RT-qPCR tests evaluated in this study can be applied to quantify viral loads ranging between 2/4 and 8 Log copy number in bees and hive debris.
4. Discussion
This study, undertaken to select the most specific and inclusive methods among those described for the detection of the most relevant honeybee viruses by RT-qPCR, led to the modification of some existing methods or design of new ones to extend the possibility to detect a higher number of virus variants. The finding that some previously reported tests were not sufficiently specific or partially inclusive is explained by the low number of target sequences available at the time when the assays were designed. This observation indicates the necessity to periodically re-evaluate molecular detection methods against recently acquired sequence data.
In this study duplex RT-qPCR reactions in the presence of an RNA extraction control for honeybee viruses were performed for the first time and gave good results in terms of sensitivity and linearity range for all targets. Therefore, the possibility to carry out different determinations in a single assay was demonstrated, with advantage for rapidity and work simplification.
The cross reactivity of the DWVA specific test towards DWVB and vice versa can be explained with the high nucleotide identity (89 %) between these two variants [De Souza 2019]. However, cross reactivity does not compromise the detection of the two variants when they are dominant in the sample. Since DWVB cannot be detected in the duplex reactions when its copy number is more than 2 Log lower than DWVA and DWVA cannot be detected in the duplex reactions when its copy number is more than 1 Log lower than DWVB, use of singleplex reactions can allow the detection of the less abundant target, similarly to the previously designed tests specific for one variant but with at least equal specificity and inclusivity.
The sensitivity reached can be sufficient to detect honeybee viruses in real samples since, for example, the predicted mean viral load for DWVB is 1.5 × 104 viral particles per bee on Varroa-free sites for A. mellifera [21].
The RT-qPCR methods described in this study showed a sufficiently wide linearity range also in detection from hive debris, thus proving useful for future non-invasive diagnosis of honeybee viral infections. The application to other hive matrices such as honey and pollen will be considered in the future since these materials were shown to better reflect the viral profile of forager bees [53].
The assays described here can further be improved by application with a recently developed semi-automated magnetic beads-based based technology applied to DWVA and DWVB RNA extraction from honeybee samples that allows the rapid screening of viral loads in 96 samples simultaneously [47].
Author Contributions
Conceptualization, F.R. and L.M.; methodology, F.R. and I.D.M.; validation, F.R., I.D.M and L.R.; formal analysis, F.R. and I.D.M.; investigation, F.R.; resources, F.R.; data curation, F.R.; writing—original draft preparation, F.R.; writing—review and editing, F.R. and L.M.; supervision, L.M.; project administration, F.R.; funding acquisition, L.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the project “IZS AM 08/19 RC. NGS e diagnostica molecolare in Sanità Animale: Fast D2” financed by the Italian Ministry of Health.
Data Availability Statement
Data can be made available upon request to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Osterman, J.; Aizen, M.A.; Biesmeijer, J.C.; Bosch, J.; Howlett, B.G.; Inouye, D.W.; Jung, C.; Martins, D.J.; Medel, R.; Pauw, A.; Seymour, C.L.; Paxton, R.J. Global Trends in the Number and Diversity of Managed Pollinator Species. Agric. Ecosyst. Environ. 2021, 322, 107653. [Google Scholar] [CrossRef]
- Beaurepaire, A.; Piot, N.; Doublet, V.; Antunez, K.; Campbell, E.; Chantawannakul, P.; Chejanovsky, N.; Gajda, A.; Heerman, M.; Panziera, D.; Smagghe, G.; Yañez, O.; de Miranda, J.R.; Dalmon, A. Diversity and Global Distribution of Viruses of the Western Honey Bee, Apis Mellifera. Insects 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Di Prisco, G.; Annoscia, D.; Margiotta, M.; Ferrara, R.; Varricchio, P.; Zanni, V.; Caprio, E.; Nazzi, F.; Pennacchio, F. A Mutualistic Symbiosis between a Parasitic Mite and a Pathogenic Virus Undermines Honey Bee Immunity and Health. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 3203–3208. [Google Scholar] [CrossRef] [PubMed]
- McMahon, D.P.; Natsopoulou, M.E.; Doublet, V.; Fürst, M.; Weging, S.; Brown, M.J.F.; Gogol-Döring, A.; Paxton, R.J. Elevated Virulence of an Emerging Viral Genotype as a Driver of Honeybee Loss. Proc. Biol. Sci. 2016, 283. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, J. Viruses In Bees: What Do They Do and What Can We Do about It? Bee World 2012, 89, 2–5. [Google Scholar] [CrossRef]
- Schurr, F.; Tison, A.; Militano, L.; Cheviron, N.; Sircoulomb, F.; Rivière, M.-P.; Ribière-Chabert, M.; Thiéry, R.; Dubois, E. Validation of Quantitative Real-Time RT-PCR Assays for the Detection of Six Honeybee Viruses. J. Virol. Methods 2019, 270, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Thu, H.T.; Thi Kim Lien, N.; Thuy Linh, M.; Le, T.H.; Hoa, N.T.; Hong Thai, P.; Reddy, K.E.; Yoo, M.S.; Kim, Y.-H.; Cho, Y.S.; Kang, S.W.; Quyen, D.V. Prevalence of bee viruses among Apis cerana populations in Vietnam. J. Apic. Res. 2016, 55, 379–385. [Google Scholar] [CrossRef]
- Hassanyar, A.K.; Huang, S.; Li, Z.; Rizwan, M.; Mehmood, S.; Raza, M.F.; Qasim, M.; Hussain, M.; Su, S. Prevalence of Bee Viruses in Apis Cerana Cerana Populations from Different Locations in the Fujian Province of China. Microbiologyopen 2019, 8, e00830. [Google Scholar] [CrossRef] [PubMed]
- Budge, G.E.; Simcock, N.K.; Holder, P.J.; Shirley, M.D.F.; Brown, M.A.; Van Weymers, P.S.M.; Evans, D.J.; Rushton, S.P. Chronic Bee Paralysis as a Serious Emerging Threat to Honey Bees. Nat. Commun. 2020, 11, 2164. [Google Scholar] [CrossRef]
- Brasesco, C.; Quintana, S.; Di Gerónimo, V.; Genchi García, M.L.; Sguazza, G.; Bravi, M.E.; Fargnoli, L.; Reynaldi, F.J.; Eguaras, M.; Maggi, M. Deformed Wing Virus Type a and b in Managed Honeybee Colonies of Argentina. Bull. Entomol. Res. 2021, 111, 100–110. [Google Scholar] [CrossRef]
- Bordin, F.; Zulian, L.; Granato, A.; Caldon, M.; Colamonico, R.; Toson, M.; Trevisan, L.; Biasion, L.; Mutinelli, F. Presence of Known and Emerging Honey Bee Pathogens in Apiaries of Veneto Region (Northeast of Italy) during Spring 2020 and 2021. Appl. Sci. 2022, 12, 2134. [Google Scholar] [CrossRef]
- Brzoskowski Chagas, D.; Liz Monteiro, F.; da Silva Barcelos, L.; Iuri Frühauf, M.; Botton, N.Y.; Ribeiro, L.C.; Silveira Becker, A.; Wolff, L.F.; Helena Saalfeld, M.; de Lima, M.; de Oliveira Hübner, S.; Fischer, G. Detection of Honey Bee Viruses in Apiaries in Southern Brazil through Two Standardized Multiplex RT-PCR. J. Apic. Res. 2022, 1–8. [Google Scholar] [CrossRef]
- Cilia, G.; Tafi, E.; Zavatta, L.; Caringi, V.; Nanetti, A. The Epidemiological Situation of the Managed Honey Bee (Apis Mellifera) Colonies in the Italian Region Emilia-Romagna. Vet. Sci. 2022, 9, 437. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, C.; Hu, T.; Li, J.; Zhou, H.; Ji, J.; Wu, J.; Kang, W.; Holmes, E.C.; Shi, W.; Xu, S. Nationwide Genomic Surveillance Reveals the Prevalence and Evolution of Honeybee Viruses in China. Microbiome 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Hulaj, B.; Granato, A.; Bordin, F.; Goga, I.; Merovci, X.; Caldon, M.; Cana, A.; Zulian, L.; Colamonico, R.; Mutinelli, F. Emergent and Known Honey Bee Pathogens through Passive Surveillance in the Republic of Kosovo. Appl. Sci. 2024, 14, 987. [Google Scholar] [CrossRef]
- Lamas, Z.S.; Chen, Y.; Evans, J.D. Case Report: Emerging Losses of Managed Honey Bee Colonies. Biology 2024, 13, 117. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, J.R.; Cordoni, G.; Budge, G. The Acute Bee Paralysis Virus-Kashmir Bee Virus-Israeli Acute Paralysis Virus Complex. J. Invertebr. Pathol. 2010, 103 Suppl 1, S30–47. [Google Scholar] [CrossRef]
- Formato, G.; Giacomelli, A.; Olivia, M.; Aubin, L.; Glick, E.; Paldi, N.; Cardeti, G.; Cersini, A.; Ciabatti, I.M.; Palazzetti, M.; Granato, A.; Mutinelli, F. First Detection of Israeli Acute Paralysis Virus (IAPV) in Italy. J. Apic. Res. 2011, 50, 176–177. [Google Scholar] [CrossRef]
- Bellucci, V.; Lucci, S.; Bianco, P.; Ubaldi, A.; Felicioli, A.; Porrini, C.; Mutinelli, F.; Battisti, S.; Spallucci, V.; Cersini, A.; Pietropaoli, M.; Formato, G. Monitoring Honey Bee Health in Five Natural Protected Areas in Italy. Vet. Ital. 2019, 55, 15–25. [Google Scholar]
- Tantillo, G.; Bottaro, M.; Di Pinto, A.; Martella, V.; Di Pinto, P.; Terio, V. Virus Infections of Honeybees Apis Mellifera. Ital. J. Food Saf. 2015, 4, 5364. [Google Scholar] [CrossRef]
- Manley, R.; Temperton, B.; Doyle, T.; Gates, D.; Hedges, S.; Boots, M.; Wilfert, L. Knock-on Community Impacts of a Novel Vector: Spillover of Emerging DWV-B from Varroa-Infested Honeybees to Wild Bumblebees. Ecol. Lett. 2019, 22, 1306–1315. [Google Scholar] [CrossRef]
- Riveros, G.; Arismendi, N.; Zapata, N.; Evans, D.; Pérez, I.; Aldea, P.; Vargas, M. Occurrence, Prevalence and Viral Load of Deformed Wing Virus Variants in Apis mellifera Colonies in Chile. J. Apic. Res. 2020, 59, 63–68. [Google Scholar] [CrossRef]
- Kevill, J.L.; Stainton, K.C.; Schroeder, D.C.; Martin, S.J. Deformed Wing Virus Variant Shift from 2010 to 2016 in Managed and Feral UK Honey Bee Colonies. Arch. Virol. 2021, 166, 2693–2702. [Google Scholar] [CrossRef] [PubMed]
- Paxton, R.J.; Schäfer, M.O.; Nazzi, F.; Zanni, V.; Annoscia, D.; Marroni, F.; Bigot, D.; Laws-Quinn, E.R.; Panziera, D.; Jenkins, C.; Shafiey, H. Epidemiology of a Major Honey Bee Pathogen, Deformed Wing Virus: Potential Worldwide Replacement of Genotype A by Genotype B. Int. J. Parasitol. Parasites Wildl. 2022, 18, 157–171. [Google Scholar] [CrossRef]
- Zhang, Z.; Villalobos, E.M.; Nikaido, S.; Martin, S.J. Seasonal Variability in the Prevalence of DWV Strains in Individual Colonies of European Honeybees in Hawaii. Insects 2024, 15, 219. [Google Scholar] [CrossRef] [PubMed]
- Doublet, V.; Oddie, M.A.Y.; Mondet, F.; Forsgren, E.; Dahle, B.; Furuseth-Hansen, E.; Williams, G.R.; De Smet, L.; Natsopoulou, M.E.; Murray, T.E.; Semberg, E.; Yañez, O.; de Graaf, D.C.; Le Conte, Y.; Neumann, P.; Rimstad, E.; Paxton, R.J.; de Miranda, J.R. Shift in Virus Composition in Honeybees (Apis Mellifera) Following Worldwide Invasion by the Parasitic Mite and Virus Vector Varroa Destructor. R. Soc. Open Sci. 2024, 11, 231529. [Google Scholar] [CrossRef] [PubMed]
- Norton, A.M.; Remnant, E.J.; Buchmann, G.; Beekman, M. Accumulation and Competition amongst Deformed Wing Virus Genotypes in Naïve Australian Honeybees Provides Insight into the Increasing Global Prevalence of Genotype B. Front. Microbiol. 2020, 11, 620. [Google Scholar] [CrossRef] [PubMed]
- Gisder, S.; Genersch, E. Direct Evidence for Infection of Varroa Destructor Mites with the Bee-Pathogenic Deformed Wing Virus Variant B - but Not Variant A - via Fluorescence-in Situ-Hybridization Analysis. J. Virol. 2021, 95. [Google Scholar] [CrossRef]
- Ryabov, E.V.; Posada-Florez, F.; Rogers, C.; Lamas, Z.S.; Evans, J.D.; Chen, Y.; Cook, S.C. The vectoring competence of the mite Varroa destructor for deformed wing virus of honey bees is dynamic and affects survival of the mite. Front Insect Sci 2022, 2, 931352. [Google Scholar] [CrossRef]
- Damayo, J.E.; McKee, R.C.; Buchmann, G.; Norton, A.M.; Ashe, A.; Remnant, E.J. Virus Replication in the Honey Bee Parasite, Varroa Destructor. J. Virol. 2023, e0114923. [Google Scholar] [CrossRef]
- Locke, B.; Forsgren, E.; Fries, I.; de Miranda, J.R. Acaricide Treatment Affects Viral Dynamics in Varroa Destructor-Infested Honey Bee Colonies via Both Host Physiology and Mite Control. Appl. Environ. Microbiol. 2012, 78, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, M.; Forzan, M.; Cilia, G.; Sagona, S.; Bortolotti, L.; Felicioli, A. First detection of replicative deformed wing virus (DWV) in Vespa velutina nigrithorax. Bull. Insectol. 2018, 71, 211–216. [Google Scholar]
- Bubnič, J.; Prešern, J.; Pietropaoli, M.; Cersini, A.; Moškrič, A.; Formato, G.; Manara, V.; Smodiš Škerl, M.I. Integrated Pest Management Strategies to Control Varroa Mites and Their Effect on Viral Loads in Honey Bee Colonies. Insects 2024, 15, 115. [Google Scholar] [CrossRef]
- Cox-Foster, D.L.; Conlan, S.; Holmes, E.C.; Palacios, G.; Evans, J.D.; Moran, N.A.; Quan, P.-L.; Briese, T.; Hornig, M.; Geiser, D.M.; Martinson, V.; vanEngelsdorp, D.; Kalkstein, A.L.; Drysdale, A.; Hui, J.; Zhai, J.; Cui, L.; Hutchison, S.K.; Simons, J.F.; Egholm, M.; Pettis, J.S.; Lipkin, W.I. A Metagenomic Survey of Microbes in Honey Bee Colony Collapse Disorder. Science 2007, 318, 283–287. [Google Scholar] [CrossRef]
- Kukielka, D.; Esperón, F.; Higes, M.; Sánchez-Vizcaíno, J.M. A Sensitive One-Step Real-Time RT-PCR Method for Detection of Deformed Wing Virus and Black Queen Cell Virus in Honeybee Apis Mellifera. J. Virol. Methods 2008, 147, 275–281. [Google Scholar] [CrossRef] [PubMed]
- McMahon, D.P.; Fürst, M.A.; Caspar, J.; Theodorou, P.; Brown, M.J.F.; Paxton, R.J. A Sting in the Spit: Widespread Cross-Infection of Multiple RNA Viruses across Wild and Managed Bees. J. Anim. Ecol. 2015, 84, 615–624. [Google Scholar] [CrossRef]
- Bradford, E.L.; Christie, C.R.; Campbell, E.M.; Bowman, A.S. A Real-Time PCR Method for Quantification of the Total and Major Variant Strains of the Deformed Wing Virus. PLoS One 2017, 12, e0190017. [Google Scholar] [CrossRef] [PubMed]
- Kevill, J.L.; Highfield, A.; Mordecai, G.J.; Martin, S.J.; Schroeder, D.C. ABC Assay: Method Development and Application to Quantify the Role of Three DWV Master Variants in Overwinter Colony Losses of European Honey Bees. Viruses 2017, 9. [Google Scholar] [CrossRef]
- Chantawannakul, P.; Ward, L.; Boonham, N.; Brown, M. A Scientific Note on the Detection of Honeybee Viruses Using Real-Time PCR (TaqMan) in Varroa Mites Collected from a Thai Honeybee (Apis Mellifera) Apiary. J. Invertebr. Pathol. 2006, 91, 69–73. [Google Scholar] [CrossRef]
- Blanchard, P.; Regnault, J.; Schurr, F.; Dubois, E.; Ribière, M. Intra-Laboratory Validation of Chronic Bee Paralysis Virus Quantitation Using an Accredited Standardised Real-Time Quantitative RT-PCR Method. J. Virol. Methods 2012, 180, 26–31. [Google Scholar] [CrossRef]
- Blanchard, P.; Guillot, S.; Antùnez, K.; Köglberger, H.; Kryger, P.; de Miranda, J.R.; Franco, S.; Chauzat, M.-P.; Thiéry, R.; Ribière, M. Development and Validation of a Real-Time Two-Step RT-qPCR TaqMan(®) Assay for Quantitation of Sacbrood Virus (SBV) and Its Application to a Field Survey of Symptomatic Honey Bee Colonies. J. Virol. Methods 2014, 197, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Šimenc, L.; Knific, T.; Toplak, I. The Comparison of Honeybee Viral Loads for Six Honeybee Viruses (ABPV, BQCV, CBPV, DWV, LSV3 and SBV) in Healthy and Clinically Affected Honeybees with TaqMan Quantitative Real-Time RT-PCR Assays. Viruses 2021, 13, 1340. [Google Scholar] [CrossRef] [PubMed]
- Castelli, L.; Genchi García, M.L.; Dalmon, A.; Arredondo, D.; Antúnez, K.; Invernizzi, C.; Reynaldi, F.J.; Le Conte, Y.; Beaurepaire, A. Intra-Colonial Viral Infections in Western Honey Bees (Apis Mellifera). Microorganisms 2021, 9, 1087. [Google Scholar] [CrossRef] [PubMed]
- Cilia, G.; Zavatta, L.; Ranalli, R.; Nanetti, A.; Bortolotti, L. Replicative Deformed Wing Virus Found in the Head of Adults from Symptomatic Commercial Bumblebee (Bombus Terrestris) Colonies. Vet. Sci. 2021, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Mráz, P.; Hýbl, M.; Kopecký, M.; Bohatá, A.; Hoštičková, I.; Šipoš, J.; Vočadlová, K.; Čurn, V. Screening of Honey Bee Pathogens in the Czech Republic and Their Prevalence in Various Habitats. Insects 2021, 12, 1051. [Google Scholar] [CrossRef] [PubMed]
- Leti Maggio, E.; Tofani, S.; Granato, A.; Formato, G.; Pietrella, G.; Conti, R.; Milito, M.; Pietropaoli, M.; Cersini, A.; Scicluna, M.T. First Description of the Occurrence of Slow Bee Paralysis Virus-1 and Deformed Wing Virus B in Apis mellifera ligustica Honeybee in Italy. Appl. Sci. 2024, 14, 626. [Google Scholar] [CrossRef]
- Nikulin, S.L.; Hesketh-Best, P.J.; Mckeown, D.A.; Spivak, M.; Schroeder, D.C. A semi-automated and high-throughput approach for the detection of honey bee viruses in bee samples. PLoS ONE 2024, 19, e0297623. [Google Scholar] [CrossRef] [PubMed]
- Tiritelli, R.; Flaminio, S.; Zavatta, L.; Ranalli, R.; Giovanetti, M.; Grasso, D.A.; Leonardi, S.; Bonforte, M.; Boni, C.B.; Cargnus, E.; Catania, R.; Coppola, F.; Di Santo, M.; Pusceddu, M.; Quaranta, M.; Bortolotti, L.; Nanetti, A.; Cilia, G. Ecological and social factors influence interspecific pathogens occurrence among bees. Sci Rep. 2024, 14, 5136. [Google Scholar] [CrossRef]
- Köppel, R.; Schum, R.; Habermacher, M.; Sester, C.; Piller, L.E.; Meissner, S.; Pietsch, K. Multiplex Real-Time PCR for the Detection of Insect DNA and Determination of Contents of Tenebrio Molitor, Locusta Migratoria and Achaeta Domestica in Food. Eur. Food Res. Technol. 2019, 245, 559–567. [Google Scholar] [CrossRef]
- Fujiyuki, T.; Takeuchi, H.; Ono, M.; Ohka, S.; Sasaki, T.; Nomoto, A.; Kubo, T. Novel Insect Picorna-like Virus Identified in the Brains of Aggressive Worker Honeybees. J. Virol. 2004, 78, 1093–1100. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; Vandesompele, J.; Wittwer, C.T. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- de Souza, F.S.; Kevill, J.L.; Correia-Oliveira, M.E.; de Carvalho, C.A.L.; Martin, S.J. Occurrence of Deformed Wing Virus Variants in the Stingless Bee Melipona Subnitida and Honey Bee Apis Mellifera Populations in Brazil. J. Gen. Virol. 2019, 100, 289–294. [Google Scholar] [CrossRef]
- Čukanová, E.; Prodělalová, J.; Palíková, M.; Kováčová, K.; Linhart, P.; Papežíková, I. Can the Examination of Different Types of Hive Samples Be a Non-Invasive Method for Detection and Quantification of Viruses in Honey Bee (Apis Mellifera L.) Colonies? J. Vet. Res. 2023, 67, 323–331. [Google Scholar] [CrossRef]
Figure 1.
Standard curves for the quantification of viruses in honeybees and hive debris in duplex RT-qPCR reactions. Red symbols, ABPV, CBPV, DWVA; blue symbols, BQCV, DWVB, SBV. Ct values are shown for the linearity range of the calibration curves.
Figure 1.
Standard curves for the quantification of viruses in honeybees and hive debris in duplex RT-qPCR reactions. Red symbols, ABPV, CBPV, DWVA; blue symbols, BQCV, DWVB, SBV. Ct values are shown for the linearity range of the calibration curves.
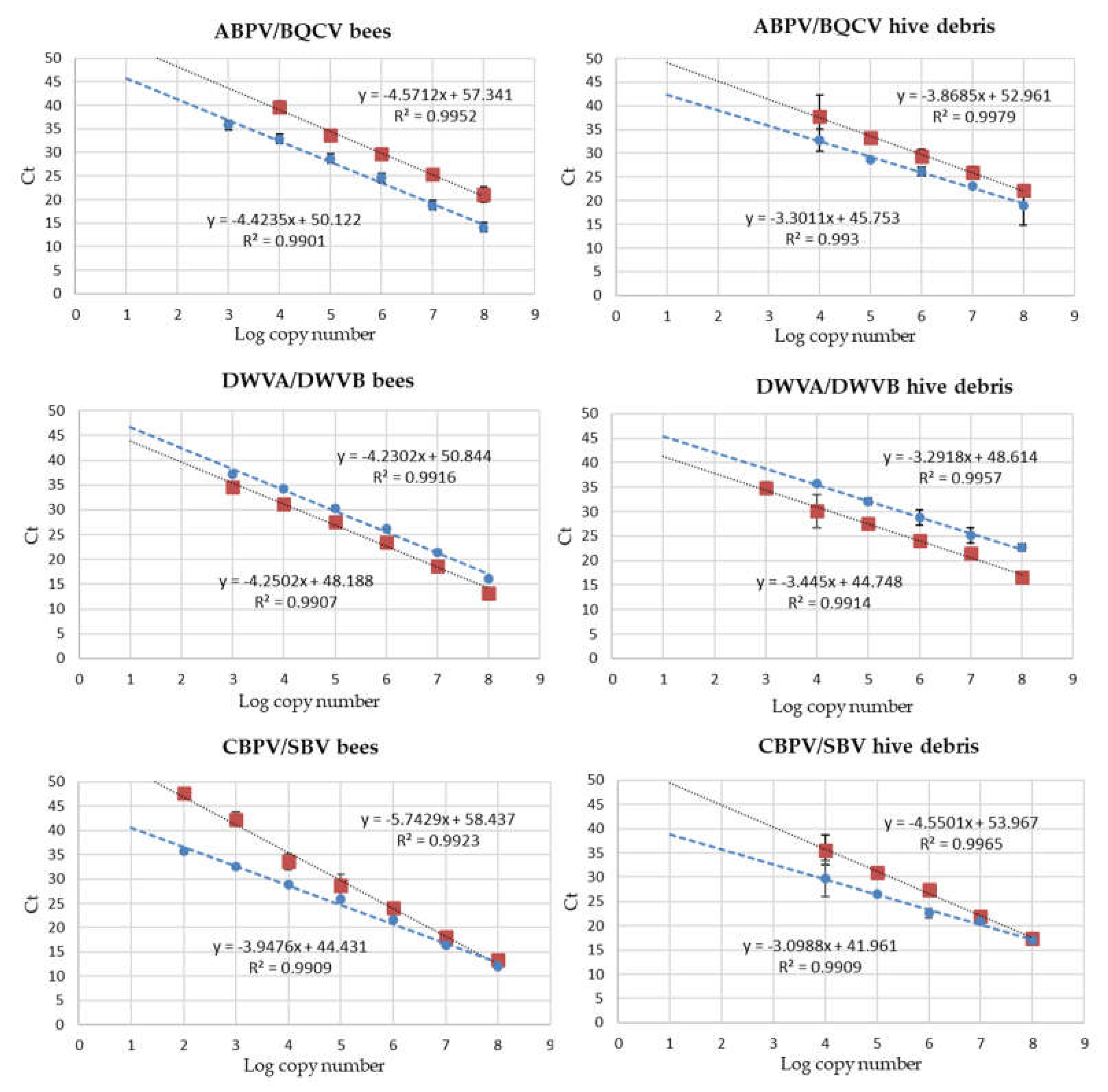

Table 1.
Sequences of primers and probes experimentally tested in this study for each honeybee virus with target genes and annealing positions on whole genome database entries.
Table 1.
Sequences of primers and probes experimentally tested in this study for each honeybee virus with target genes and annealing positions on whole genome database entries.
| Label | Sequence 5′-3′* | Target | Nucleotide positions |
|---|---|---|---|
| Acute paralysis virus (ABPV) | |||
| APVF APVP APVR |
TTTGTTTCAAAYAARATGTTYATGAAAYC FAM-TATGGTGGAAAYKCTGARAAYAAT-MGBEQ† BTWGAHACAGTCTCTGGACACAT |
Capsid protein gene | Acc. n. ON648748.1 8324-8466 |
| Black queen cell virus (BQCV) | |||
| BQCVF BQCVP BQCVR |
GTGCGGGAGATGATATGGA Cy5-TTTCCATCTTTATCGGTACGC-MGBEQ CCGTCTGAGATGCATGAATAC |
Capsid protein gene | Acc. n. MT482476.1 8060-8128 |
| Chronic bee paralysis virus (CBPV) | |||
| CBPVF CBPVP CBPVR |
GAAGTCATCCGTAGATCTGG FAM-AGACKAGRGAGGAYGGGA-MGBEQ CRAGAGGGGTATGTTGTACT |
RNA1 gene | Acc. n. MK637522.1 1961-2070 |
| Deformed wing virus A (DWVA) | |||
| DWVAF DWVAP DWVAR |
CTTTGTCTTCATTAAAGCCAC FAM-TGCGTGGAATGCGTCC-MGBEQ CTCATTAACTGTGTCGTTGAT |
Polyprotein gene | Acc. n. OR497397.1 8636-8774 |
| Deformed wing virus B (DWVB) | |||
| DWVBF DWVBP DWVBR |
TTTATCTTCATTAAAACCGCCA Cy5-ATCTTTTGAGAGGGATGAGA-MGBEQ CTCATTAACTGAGTTGTTGTC |
Polyprotein gene | Acc. n. OR497394.1 8615-8752 |
| Sacbrood virus (SBV) | |||
| SBVF SBVP SBVR |
AAYGTCCACTACACCGAAATGT Cy5-TGATGAGAGTGGACGAAGAATCTGGAATG-BHQ2 TAHGAGGTAATAACTTTTCGCCA |
Polyprotein gene | Acc. n. MN082652.1 430-548 |
* degenerate nucleotide position code: H (A,C,T); K (G,T); Y(C,T); R (A,G) (https://www.bioinformatics.org/sms/iupac.html, accessed on 24 April 2024).
Table 2.
P values obtained by the Student’s t test in the comparison of Ct values obtained in triplicate reactions containing one or both the synthetic target RNAs.
Table 2.
P values obtained by the Student’s t test in the comparison of Ct values obtained in triplicate reactions containing one or both the synthetic target RNAs.
| Log copy number | ||||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| Target | P values | |||||
| ABPV | 0.91 | 0.90 | 0.98 | 0.92 | 0.31 | 0.33 |
| BQCV | 0.90 | 0.87 | 0.51 | 0.96 | 0.53 | 0.10 |
| CBPV | 0.98 | 0.05 | 0.22 | 0.72 | 0.15 | 0.58 |
| DWVA | 0.02 | 0.44 | 0.97 | 0.78 | 0.08 | 0.37 |
| DWVB | 0.16 | 0.18 | 0.48 | 0.75 | 0.15 | 0.57 |
| SBV | 0.51 | 0.80 | 0.79 | 0.28 | 0.91 | 0.65 |
Bold character: statistically significant for P<0.05.
Table 3.
LOD values for each viral target in the duplex RT-qPCR tests carried out on solutions of the synthetic RNA targets and on RNA extracts from honeybees and hive debris samples spiked with the synthetic RNA targets.
Table 3.
LOD values for each viral target in the duplex RT-qPCR tests carried out on solutions of the synthetic RNA targets and on RNA extracts from honeybees and hive debris samples spiked with the synthetic RNA targets.
| Log copy number | |||
| PCR reaction | One bee | 100 µl hive debris* | |
| Target | |||
| ABPV | 1.99 | 3.65 | 3.47 |
| BQCV | 1.37 | 2.81 | 2.88 |
| CBPV | 1.21 | 2.9 | 2.72 |
| DWVA | 1.25 | 2.95 | 2.77 |
| DWVB | 1.39 | 4.07 | 3.90 |
| SBV | 1.49 | 3.17 | 4.00 |
*approximate volume of hive debris.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.


Selection and Multiplexing of Reverse Transcription Quantitative PCR Tests Targeting Relevant Honeybee Viral Pathogens
Franca Rossi
et al.
,
2024
RT-qPCR Diagnostics: The “Drosten” Sars-Cov-2 Assay Paradigm
Stephen Bustin
et al.
,
2021



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated