
Preprint
Article
Of SINEs, LINEs and Leeches: Identification and Molecular Characterization of Transposable Elements in Leech Genomes
Altmetrics
Downloads
101
Views
28
Comments
0
supplementary.pdf (264.30KB )


This version is not peer-reviewed
Submitted:
02 August 2024
Posted:
06 August 2024
You are already at the latest version
Alerts
Abstract
Mobile genetic elements constitute a major part of almost every eukaryotic genome, and several types of such elements have been classified based on size, genetic structure and transposition intermediate. The fast growing availability of whole genome sequences of species across the living world provides almost unlimited possibilities for in depth molecular analyses of all kind, including the search for mobile genetic elements in animal taxa that have not yet been in the focus of respective investigations. Leeches are such a group of so far neglected organisms. However, aim of the present study was not to provide a comprehensive survey, but to perform the first molecular description and characterization of selected mobile genetic elements (MGEs) in leeches. Representatives of three types of MGEs could be identified and characterized, namely short interspersed nuclear elements (SINEs), long interspersed nuclear elements (LINEs) and long terminal repeats (LTR)-retro-transposons. Some of the newly described elements display unique structural features compared to archetype elements of the respective groups.

Keywords:
Subject: Biology and Life Sciences - Biochemistry and Molecular Biology
Introduction
Mobile genetic elements (MGEs) or
transposable elements (TEs) are present in almost every complex eukaryotic
genome and comprise up to 20 % of the total genome size in fungi, up to 50 % in
metazoans and up to 90 % in plants (Wicker et al. 2007). TEs hence not only
shape the structure of a genome, but may also change it due to their ability to
move and replicate. As a consequence, TEs contribute to genomic plasticity and
are major drivers of genome evolution (Kazazian 2004). As such their mobility
may result in beneficial (gain or enhancement of traits) or detrimental (loss
or change of traits, disease development) outcomes for the affected cell and the
whole individual.
TEs have been classified according
to their transposition intermediate (RNA or DNA) as class I elements (or
retrotransposons) and class II elements (or DNA transposons) (Finnegan 1989).
Later Wicker et al. (2007) introduced an unified classification system for
eukaryotic TEs that includes
subclasses, orders,
superfamilies, families and subfamilies. As an example, the class I elements
can be further subdivided based on the presence or absence of long terminal
repeats (LTRs) to form the subclasses of either LTR- or
Non-LTR-retrotransposons. Among the latter, respective elements may either
transpose autonomously or non-autonomously depending on whether or not they
encode for proteins that catalyze the retrotransposition event (Kazazian 2004;
Finnegan 2012). Autonomous Non-LTR-retrotransposons enclose the superfamiliy of
long interspersed nuclear elements (LINEs), whereas the superfamily of short
interspersed nuclear elements (SINEs) transpose non-autonomously and depend on
the trans activity of their respective LINE counterparts for mobility
(Wells and Feschotte 2020; Bourque et al. 2018). For that SINEs have to share a
3`end sequence with their corresponding LINE. The shared sequence can be very
specific to form a stringent SINE/LINE pair or can be an unspecific polyA tail
to form a relaxed SINE/LINE pair (Okada et al. 1997; Roy-Engel 2012).
PolyA tails are usually generated during mRNA synthesis by the RNA
polymerase II (pol II), but can also be generated by RNA polymerase III (pol
III) during the transcription of respective SINEs. The latter process requires
the presence of a polyadenylation signal (pAS, AATAAA) and a pol III terminator
sequence (TCTTT) within the SINE sequence (Borodulina and Kramerov 2008;
Roy-Engel 2012). Respective SINEs belong to the T+-class, whereas
SINEs lacking both sequence signals belong to the T--class and
already contain an A-rich tail (Borodulina and Kramerov 2001). Both T+-
and T--class SINEs can hence “cooperate” with various LINEs as long
as the respective LINE encodes a polyA tail by itself.
LINEs are about 3 - 6 kb in size
and comprise one or more open reading frame(s) (ORF) that code for at least two
proteins that facilitate reverse transcription and transposition, a reverse
transcriptase (RT) and an endonuclease, but additional domains like for a RNAse
H (RH) might be present. Presence and arrangement of the respective domains
form the basis for LINE classification (Wicker et al. 2007). In addition to the
domains several sequence motifs are frequently present in LINE-encoded proteins
including the CCHC zinc finger knuckle and a RNA-recognition motif (RRM)
(Khazina and Weichenrieder 2007; Metcalfe and Casane 2014). A full RRM motif in
turn is composed of two short motifs known as ribonucleoprotein (RNP) motifs
RNP1 and RNP2 (SenGupta 2013). SINEs in contrast are much smaller (appr. 80 -
500 bp in size) (Wicker et al. 2007; Kramerov and Vassetzky 2011) and are
composed of a head structure that contains a pol III promoter region, a central
core domain and a LINE-related segment (Gilbert and Labuda 1999). The SINE
promoters can be derived from either tRNA, 5S or 7SL genes, respectively
(Kramerov and Vassetzky 2011). As a basic rule, both LINEs and SINEs generate
target site duplications (TSDs) of variable length upon insertion that can be
used to annotate and classify the respective element (Li et al. 2022; Goubert
et al. 2022), but TSDs may mutate and degrade over time (Kanhayuwa and Coutts
2016).
Leeches belong to the phylum Annelida
(segmented worms) and the class Clitellata. They are globally
distributed with exception of Antarctica, and about 700 leech species have been
described so far (Sket and Trontelj 2008). However, the actual diversity of
leeches might be much higher (Kvist et al. 2022). Whereas some leeches are
predators, others are hematophagous and require regular blood meals for growth,
development and reproduction (Sawyer 1986). Probably the most famous of all
leeches is Hirudo medicinalis Linnaeus, 1758, the (Middle) European
medicinal leech, but leeches have been used for medical purposes for thousands
of years in many cultures worldwide (Abdualkader et al. 2013). To ensure an
undisturbed and saturating blood meal, leeches secrete a great variety of bioactive
substances into the bite, among them factors that interfere with the
coagulation cascade, inhibit inflammation or prevent pain sensation
(Hildebrandt and Lemke 2011; Lemke and Vilcinskas 2020). Despite the great
biopharmaceutical potential, the thrombin-inhibitor hirudin is the only
leech-derived compound that found its way from nature to clinical application
(Greinacher and Warkentin 2008). Over the last years, the whole genome
sequences of a few leech species have been determined, namely Helobdella
robusta Shankland, Bissen and Weisblat, 1992 (Simakov et al. 2013), Hirudinaria
manillensis Lesson, 1842 (Guan et al. 2020), H. medicinalis (Kvist
et al. 2020; Babenko et al. 2020), Whitmania pigra Blanchard, 1887 (Tong
et al. 2022) and Hirudo verbana Carena, 1820 (Paulsen et al. 2020,
version 2023, submitted). A detailed analysis of the H. medicinalis and H.manillensis
genomes using the RepeatMasker and RepeatModeler pipelines revealed the
presence of a variety of putative TEs including DNA transposons, LTR-retrotransposons,
LINEs and SINEs with copy numbers between 3 and several thousand for each
particular element (supplementary information 1 in Kvist et al. 2020; Supplementary
Material table S2 in Guan et al. 2022). According to Zhao et al. (2024) about
30 % of the total genome size of Hirudo nipponia Whitmann, 1886, and Hirudo
tianjinensis Liu, sp. nov., are formed by repeat elements. However, a more
detailed analysis and in-depth characterization of leech-derived putative TEs
is missing. In the present study the author describes the identification and
molecular characterization of SINEs that transposed into hirudin genes of H.
verbana and H. manillensis and analyze their relationship to
respective TEs (SINEs, LINEs and LTR-elements) of different leech taxa.
Methods and Materials
Genome and Transcriptome Data
Leech genome data for
H. robusta
, H. manillensis, H.
medicinalis , W. pigra and H. verbana are freely accessible
and searchable through public databases. Available transcriptome data were used
to complement the genome-based investigations when necessary.
Sources of Reference Sequences
The following references were used
to identify the signatures of TE-related domains and motifs:
| Domain | Reference |
| Apurinic endonuclease (APE) | Fillingham et al. 2004; Kojima and Fujiwara 2005 |
| Aspartic protease (AP) | Tözsér 2010; Gazda et al. 2020 |
| Integrase (IN) | Evgen`ev et al. 1997; Ohta et al. 2002 |
| Restriction-like endonuclease (RLE) | Kojima and Fujiwara 2005 |
| Reverse Transkriptase (RT) | Evgen`ev et al. 1997; Goodwin and Poulter 2001; Arkhipova 2006; Meier et al. 2006 |
| RNA recognition motif (RRM) | Maris et al. 2005; Khazina and Weichenrieder 2009 |
| RNAse H (RH) | Lingner et al. 1997; Xu et al. 2016; Moelling et al. 2017 |
| Tyrosin recombinase (YR) | Goodwin and Poulter 2004; Poulter and Goodwin 2005; Poulter and Butler 2015 |
| Zinc finger knuckle motif (CCHC) | Krishna et al. 2003 |
Bioinformatics Tools
Basic Local Alignment Search Tool
(BLAST) searches were performed using the respective NCBI web portal and
default settings for search algorithms parameters.
Multiple sequence alignments were
generated
using the CLC Sequence Viewer software
package v8.0 (CLC bio) and default settings.
Phylogenetic trees were generated
using the
CLC Sequence Viewer software package v8.0,
the UPGMA algorithm and default settings.
Putative TSDs were identified
using the Web-based tool “Repeats Finder for DNA/Protein Sequences” (https://www.novoprolabs.com/tools/repeats-sequences-finder).
Results
Identification and Characterization of HvSINE1
In previous studies we have
determined the gene structures of several hirudin and hirudin-like factor (HLF)
genes including hirudin-variants HV1, HV2 and HV3 of Hirudo medicinalis
(Müller et al. 2016) and Hirudo verbana (Müller et al. 2017). In all
cases the genes shared a highly conserved structure not only in exon and intron
number, but also in terms of position and size. Our findings were confirmed
upon the availability of whole genome data of H. medicinalis (Kvist et
al. 2020; Babenko et al. 2020). Only recently whole genome data of H.
verbana became accessible via GenBank (BioProject
PRJNA55103,
Sequence Read Archive SRS5059564), the respective
manuscript was deposited at BioRxiv and is currently under review (Paulsen et
al. 2020, version 2023, submitted). Surprisingly, a detailed analysis revealed
remarkable differences in the structure of the HV1 gene of the particular H.
verbana biosample that was used for the study of Paulsen et al. (2020/2023)
compared to both the biosamples of our own studies (GenBank accession numbers
KX215734.1 and KX215735.1 for H. verbana and KR066930.1 and KR066931.1
for H. medicinalis) and the investigations of Kvist et al. (2020) and
Babenko et al. (2020). The sizes of introns 2 and 3 differ by 210 or 25 bp,
respectively (Table 1).
Whereas the alterations in intron
3 are scattered across the element, a multiple sequence alignment reveals the
additional presence of a continuous sequence stretch in intron 2 (Figure 1, marked in cyan in
Supplementary Material Figure S1).
To evaluate, whether or not the
extra DNA may represent a putative TE the author performed a BLAST search
against the whole genomes of both H. verbana and H. medicinalis.
In both cases, the number of hits by far exceeded the display options,
indicating the presence of thousands of copies of the respective element in the
genomes of both leech species. The extra DNA hence most likely indeed
represents a putative TE. The best 50 hits within the genome of H. verbana
were extracted and aligned to determine the consensus sequence of the putative
TE (Supplementary Material Figure S2). Strikingly,
no evidence for target site duplications (TSDs) could be found.
To decipher to what class of TEs
the element might belong the author first tested the hypothesis that it
represents a SINE. As already outlined in the Introduction section, SINEs
comprise a conserved overall structure, but differ in their promoter origin
(tRNA, 5S or 7SL genes). The respective promoter sequences of H. verbana
or H. medicinalis were determined and used to assign the putative TE.
The predicted promoter sequence does not perfectly match to one or the other
archetype sequences, but structure (Box1 and
Box 2),
sequence and spacing between the boxes strongly point to a tRNA gene derived
promoter (Figure 2).
Identification and Characterization of HvSINE2-4
Based on the core domain sequence of HvSINE1, additional BLAST searches were performed and revealed evidence for the presence of related SINEs in H. verbana. The respective elements were termed HvSINE2-4. The four SINEs show overall degrees of sequence similarity between 35 and 77 % with HvSINE4 being the most distinct member of the family (Supplementary Material Figure S3). All four elements contain tRNA gene derived promoters (Figure 2C), share a common core domain of 55 bp in size (underlined in Figure 3), but differ in their LINE-specific segments. Strikingly, only HvSINE4 contains a short repeat sequences at the 3`end (Figure 3), an otherwise typical feature of SINEs (Gilbert and Labuda 1999).
The abundances of HvSINE1-4 differ markedly in the genomes of both H. verbana and H. medicinalis: whereas HvSINE1 is present in very high numbers, HvSINE2 and HvSINE3 each occur in a single copy only. For HvSINE4, 21 copies in H. verbana and 14 copies in H. medicinalis comprise the whole sequence, whereas about 200 copies in each genome contain the head and core domains, but lack the LINE-specific segment (Table 2).
Tissue-Specific Expression of HvSINE Sequences
Like all retrotransposons, SINEs transpose via a “copy-paste-mechanism” including transcription of the element (Kramerov and Vassetzky 2011). It should hence be possible to detect the respective SINE sequences in transcriptome datasets as well. Several tissue-specific transcriptome datasets of either H. verbana or H. medicinalis including muscle (SRX3875125), salivary gland (SRX3875124), central nervous system (CNS) (SRX3742574), ganglion (SRX9699081, SRX9699082, SRX9699083) and head (SRX5257616) were analyzed. Both HvSINE1 and HvSINE4 sequences could be detected in all datasets, whereas the expression of HvSINE2 and HvSINE3 seems to be restricted to neuronal tissue (Table 3).
Presence of HvSINE1-Like Elements in Other Leeches
To evaluate the presence of HvSINE1-like elements in leeches outside the genus Hirudo the author performed BLAST searches in genome and/or transcriptome datasets of various leech and annelid species. The results are summarized in Table 4 and clearly indicate that the presence of HvSINE1-like elements is restricted to merely a handful of Eurasian members of the family Hirudinidae. Among them are two non-hematophagous leeches, namely Haemopis sanguisuga Linnaeus, 1758, and Whitmania pigra. As for H. verbana and H. medicinalis, for each leech species several distinct SINEs could be identified. The sequence data for all elements are summarized in Supplementary Material Figures S4 (Hirudinaria manillensis), S5 (W. pigra), S6 (H. sanguisuga) and S7 (Hirudo nipponia).
Phylogenetic Analyses Based on the HvSINE1 Sequence
The presence of SINEs in almost all vertebrate and invertebrate taxa make them promising candidates as markers for molecular phylogeny and systematics (Miyamoto 1999; Ray et al. 2006; Korstian et al. 2022). To get an impression whether or not leech SINEs might be useful tools for phylogenetic analyses as well the author constructed trees based on either cytochrome C subunit I (coi) sequences that are commonly used for DNA barcoding or on HvSINE1-like sequences. Best matches to HvSINE1 in every leech species were selected and included into the analysis. The coi-sequence of Lumbricus terrestris Linnaeus, 1758, and the sequence of HvSINE4 were choosen as outgroups for the respective trees. The resulting trees were manually redrawn to illustrate the basic principles, not the actual distances. As can be seen in Figure 4, the trees match well, but not perfectly to each other. Nevertheless SINEs can seriously be considered as additional molecular markers for phylogenetic analyses in leeches.
Original trees were redrawn and branch lengths do not represent phylogenetic distances. Lter: Lumbricus terrestris: Hver: Hirudo verbana; Hmed: Hirudo medicinalis; Hman: Hirudinaria manillensis; Hsan: Haemopis sanguisuga; Wpig: Whitmania pigra; Hnip: Hirudo nipponia; Hver1: sequence of HvSINE1; Hver4: sequence of HvSINE4
Identification and Characterization of HmSINE_V2
In a recent manuscript we described the identification of a Tandem-hirudin (TH) including the corresponding gene in H. manillensis (Lukas et al. 2022). In contrast to the archetype hirudin gene, the TH gene is composed of 6 exons and 5 introns. Within the fifth exon (565 bp in size), a quite unusual stretch of 18 thymine residues giving rise to a polyA tail in the reverse-complementary orientation caught the attention. As already mentioned in the Introduction section, SINEs of the T--class contain an A-rich tail, and a thorough analysis of TH gene exon 5 indeed revealed strong evidence for the presence of yet another SINE in H. manilesis. The putative TE is very different from the HvSINE1-like elements described above and was termed HmSINE_V2, its sequence is given in Figure 5. About 50 copies of HmSINE_V2 are present in the genome of H. manillensis. Interestingly, searches against the genomes of H. verbana/medicinalis and W. pigra revealed the presence of similar elements that, however, did not cover the entire sequence of HmSINE_V2, but started only at position 151 (the respective matching sequence is underlined in Figure 5). Another quite curious aspect of HmSINE_V2 is the presence of both a pAS and pol III terminator sequence in addition to the A-rich tail. The element hence comprises features of both T+- and T--class SINEs.
Identification of Corresponding LINEs
As already outlined in the Introduction section, SINEs are non-autonomous TEs and depend on the trans activity of their respective LINE counterparts for mobility. In other words: Where`s a SINE, there`s a LINE. Despite extensive efforts the author failed to identify the corresponding LINEs for HvSINEs1-3. However, for both HvSINE4 and HmSINE_V2 putative matching LINEs could be identified. The elements were termed HvLINE1 and HmLINE1, respectively; the sequences (both nucleotide and derived amino acid sequences of predicted open reading frames (ORFs)) are given in Supplementary Material Figures S8 and S9. The elements are app. 4.4 kb (HvLINE1) and 3.5 kb (HmLINE1) in size. HmLINE1 is flanked by a putative 8 bp TSD and contains a single ORF that encodes a protein of 946 amino acid residues in length. Compared to HmLINE1, the structure of HvLINE1 is rather complex. First, no putative TSD could be determined. Second, the element contains four ORFs, the first in reverse-complementary orientation. The structures of both elements are represented in Figure 6 as schematic drawings. HvSINE4 and HvLINE1 share a stretch of 48 bp in length (double underlined in Figure 3 and Supplementary Material Figure S10), a typical size for LINE-related segments in SINEs (Gilbert and Labuda 1999). In contrast, the entire sequence of HmSINE_V2 is present in HmLINE1 including the stop codon of the ORF (Supplementary Material Figure S10).
Both HvLINE1 and HmLINE1 encode AP, RT and RH domains and a CCHC motif, respectively, but only HvLINE1 encodes a full RRM. Strikingly, the AP domain and the two RNP motifs of HvLINE1 are encoded by different ORFs (ORF1 and 2) that are orientated in opposite direction to each other (see Figure 6).
Further analyses led to the identification of additional putative LINEs in H. verbana, named HvLIN2-4. The nucleotide and amino acid sequences of HvLINEs2-4 are provided in Supplementary Material figures S11-S13. All four LINEs of H. verbana display different structures in terms of overall size, ORF number and size, presence and localization of CCHC and RRM motifs and presence of TSDs (Figure 6), highlighting the great diversity of such TEs even within a single species.
For HmLINE1, analyses revealed the presence of a similar element in W. pigra, named WpLINE1. In contrast to HmLINE1, WpLINE1 contains not only one, but four ORFs. However, most likely the ORFs1-3 belong to a putative LTR-retrotransposons (named WpLTRE1) that integrated into WpLINE1, disrupting the “original” single ORF of WpLINE1 and creating a patchwork element (Figure 6). WpLTRE1 itself is flanked by direct repeats of 105 bp in size (highlighted in purple in Figure 6 and Supplementary Material Figure S14). When eliminating WpLTRE1 and manually reconstructing the ORF of WpLINE1, the elements encodes two putative APE domains (Figure 6 and Supplementary Material Figure S14). Both HmLINE1 and WpLINE1 contain an A-rich tail immediately downstream of the pol III terminator sequence (Supplementary Material Figures S9 and S14).
Abundance of WpLTRs
The entire sequence of WpLTRE1 including the LTRs was used to evaluate, whether or not additional copies of the element are present in the genome of W. pigra. It turned out that indeed additional copies are present. All sequences are of about the same size of app. 2.2 kb and comprise the full length LTRs, confirming that they are integral part of WpLTRE1. No target sequence duplications and no evidence for target site specificity for integration could be observed. The sequences of the “original” WpLTRE1 (named copy 1) and four additional copies (named copy 2-5) including a multiple sequence alignment of the deduced amino acid sequences are shown in Supplementary Material Figure S15. Strikingly, all copies of WpLTRE1 but copy 1 (the “original”) contained a single ORF only, spanning almost the full length of the element and comprising the three ORFs of copy 1 (see Supplementary Material Figure S15). Hence copy 1 most likely represents an atypical WpLTRE1 and copies 2-5 likely represent the archetype WpLTRE1. The deduced amino acid sequences of all copies reveal an interesting feature of WpLTRE1. The WpLTR1copies 2-5 encode a protein of 637 amino acid residues in length that encompasses a putative YR domain including the highly conserved RHRY tetrad, a CCHC motif, a putative AP domain as well as a putative IN domain including the highly conserved DDE(K) triad (see Figure 6 and Supplementary Material Figure S15). In WpLTR1 copy 1, the three domains are encoded by the three ORFs (Figure 6). The YR domain is a characteristic feature of the DIRS1 group of retrotransposons (Goodwin and Poulter 2001), a group that does not contain LTRs, whereas the integrase domain is part of LTR-retrotransposons-encoded proteins (Haren et al. 1999). Hence WpLTRE1 combines features of both types of TEs, a phenomenon that raises questions about its actual mode of transposition.
WpLTRE1 copy 2 was used as the query sequence for a BLAST search to address the question whether or not WpLTRE1-like elements are also present in H. verbana. The search revealed the presence of several copies of a similar element, named HvLTR1. The element is flanked by LTRs of 186 bp in size and also the overall length (about 4.1 kb) is larger compared to WpLTR1 (about 2.2 kb). The sequences of two copies of HvLTR1 are presented in Supplementary Material Figure S16. HvLTR1 copy 1 and 2 differ in the length of the 5`LTR (with respect to the orientation of the ORF) due to an internal deletion of 43 bp in HvLTR1 copy 2. Both HvLTR1 sequences contain a stretch of unresolved nucleotides at the same position. Unfortunately the problem is apparent in all contig sequences that cover the whole length of the element.
Like WpLTR1 copies 2-5, HvLTR1 contains a large single ORF, however, the presence of undetermined nucleotides made any further conclusions extremely uncertain. For that reason the author tried to fill the gap and eventually identified a contig that covered the “region of uncertainty” including the flanking regions on both sides. Based on that contig the stretch of undetermined nucleotides comprises 118 bp in total. The reconstructed sequence still contains a single ORF that encodes for a protein of 1188 amino acid residues in length. The reconstructed region of 39 amino acid residues almost perfectly fits both in size and sequence to hypothetical proteins of Caenorhabditis brenneri Sudhaus and Kiontke, 2007, and Ancylostoma ceylanicum Looss, 1911 (see Supplementary Material Figure S16) making it very likely that the reconstruction was correct.
The hypothetical proteins encoded by HvLTR1 is in part highly homologous to its WpLTR1 encoded counterpart. The first 444/446 amino acid residues of both proteins display degrees of 85%/93% of sequence identity/similarity including the putative YR domain, the CCHC motif and the AP domain, but lacking the putative IN domain. The remaining parts differ markedly, however, also the hypothetical protein of HvLTR1 comprises a putative IN domain close to the C-terminus (see Supplementary Material Figure S16). Both putative IN domains differ in sequence, but localization and spacing of the canonical DDE(K) motifs are comparable. In addition, the HvLTR1 encoded protein contains both a putative RH domain and a RT domain, but as for WpLTR1, no evidence for a Gag-encoding ORF could be found.
Discussion
The number of described TEs growths constantly, and the classification system becomes increasingly complex (Arkhipova 2017; Kojima 2019). Many of these elements have been identified in “model organisms” like Drosophila (McCullers and Steiniger 2017) or Arabidopsis (Quesneville 2020), but the progress in sequencing technology and assembly methods allow for the rapid and cost-effective determination of whole genome sequences of all kinds of organisms. The respective datasets can subsequently be used to address (beside many others) questions on the presence of TEs and their impact on biological processes (e.g. Han et al. 2021). However, both the correct annotation and characterization of putative TEs in genome sequence datasets are challenging and time-consuming tasks. Tools like the RepeatMasker (Tarailo-Graovac and Chen 2009), RepeatModeler (Flynn et al. 2019) and Generic Repeat Finder (Shi and Liang 2019) pipelines provide first information, but more targeted analyses (Bell et al. 2022; Li et al. 2022) and even a final manual editing (or “curation”) of output results (Goubert et al. 2022) are mandatory. The present study did not consequently follow the pathway outlined above, but started with an accidental finding: the presence of a putative TE in a gene in H. verbana that encodes the well known leech-derived bioactive factor hirudin. The TEs that were identified and characterized in the course of the present study belong to non-LTR (SINEs and LINEs) and LTR-retrotransposons. In all cases they represent the first TEs that have been described in detail in leeches so far.
Classification of SINEs
SINEs were identified in all together six leech species, namely H. verbana, H. medicinalis, H. nipponia, H. sanguisuga, H. manillensis and W. pigra. Based on a promoter sequence determination they very likely all belong to the tRNA head superfamily (Wicker et al. 2007), but the core sequences markedly differ between the HvSINE1-3 group on one hand and HvSINE4 on the other hand (Figure 3 and Supplementary Material Figure S3). HvSINE1-like elements can be found in closely related Eurasian species of the leech family Hirudinidae, but not in family members of other geographical origin or in representatives of other leech families (Table 4). Leech SINEs may hence be used as an accessory molecular marker for phylogenetic and phylogeographic analyses, and a first sequence-based attempt to verify this hypothesis yielded convincing results (Figure 4). However, since SINEs are very short genetic elements, the actual information content of a single element is rather limited, and only the combination of various elements into one analysis may result in reliable conclusions (Deragon and Zhang 2006). An even more robust phylogenetic marker is the presence/absence pattern of SINE insertions (Nikaido et al. 1999; Korstian et al. 2022). The origin of the present study nicely illustrates the putative pitfalls of the latter strategy: the H. verbana individuals that were used in the studies by Müller et al. (2017; H. verbana_HGW) and Paulsen et al. (2020, version 2023, submitted; H. verbana_USA) display 99,5 % of coi sequence identity and both belong to the Eastern subgroup of H. verbana (Trontelj and Utevsky 2012). Integration of HvSINE1 into the hirudin HV1 gene of H. verbana_USA must hence be a very recent event, and HvSINE1 is very likely still an active TE. The latter assumption is strongly supported by its very high abundance in the genomes of both H. verbana and H. medicinalis (Table 2). Interestingly, the expression pattern of all HvSINE elements in different organs/tissues of H. verbana/medicinalis is not uniform but displays remarkable differences (Table 3). To the knowledge of the author this is the first example of such investigations, and the data are much to tentative to draw any further conclusions but may illustrate the need to pay attention on tissue-specific expression patterns of TEs in the future.
Unfortunately, for HvSIN1-3 no corresponding LINE could be identified. The very high copy number of HvSINE1 in combination with the short LINE-related segment impeded all BLAST search attempts. In contrast, for HvSINE4 a corresponding LINE, named HvLINE1, could be identified. HvSINE4 and HvLINE1 share a common segment of 48 bp in length including a short A-rich tail and a stretch of simple repeats (Supplementary Material Figure S10). The further properties of HvLINE1 will be discussed below.
A second SINE that was identified based on its presence in a hirudin gene is HmSINE_V2 in H. manillensis. Whereas all HvSINE1-like elements and HvSINE4 do not contain a pAS and a pol III terminator sequence and belong to the T--class, HmSINE_V2 does and hence belongs to the T+-class (Roy-Engel 2012). In addition the element also contains a long A-rich tail, a typical structure of T--class elements, making it a hybrid element. The most striking feature of HmSINE_V2, however, is its relationship with the respective LINE: the complete sequence of HmSINE_V2 is present in HmLINE1 (Supplementary Material Figure S10). The LINE-related segment usually comprises only a part of the entire SINE sequence located at the 3`end (Gilbert and Labuda 1999). Nevertheless, the presence of app. 50 copies of HmSINE_V2 in the genome of H. manillensis indicates that the element is not an artefact, but very likely functional.
Classification of LINEs
Six new LINEs have been identified and characterized in the course of the present investigations. Two of them, HvLINE1 and HmLINE1, could so far be attributed to respective SINES (see above). Only HvLINE2 displays the “classical” architecture of a LINE encompassing two ORFs: the first encoding a basic protein (pI value 9.41) including three CCHC motifs and the second encoding a multi-domain protein with putative domains for APE, RT and RH and a single C-terminal CCHC motif (Figure 6 and Supplementary Material Figure S11). Presence and order of domains permit a classification to either the L1 group or the I group (Kojima 2019). HvLINE3 is in large parts comparable to HvLINE2, with exception of the first ORF that is split into two separate ORFs (Figure 6 and Supplementary Material Figure S12). Very likely the proteins that are encoded by both ORFs form a heterodimer: the molecular mass of the putative heterodimer (44.7 kDa) is almost identical to the molecular mass of the protein encoded by ORF1 of HvLINE1 (44.8 kDa).
HvLINE4 belongs to a different superfamily of LINEs. The element comprises a RLE domain instead of an APE domain, a feature typical for the so called “early branched non-LTR retrotransposons” (Kojima and Fujiwara 2005). Elements of that type belong to the R2 group and usually contain a single ORF that encodes a RH domain and a RLE domain (Kojima 2019). Strikingly, HvLINE4 encodes two putative RH domains in addition to the RLE domain (Figure 6 and Supplementary Material Figure S13). To the knowledge of the author no such duplication has been described so far in LINEs.
HvLINE1 comprises a rather complex structure. The element is composed of four ORFs, the first being in opposite direction to ORFs 2-4. So far no structure like this has been described for LINEs. The assumption that ORF1 is an integral part of the element is supported by the observation that the two RNP motifs of a complete RRM and a putative APE domain are encoded by both ORF 1 and 2 (Figure 6 and Supplementary Material Figure S8). In general, the domain order of HvLINE1 resembles that of HvLINEs2 and 3, and the four proteins that are encoded by ORFs1-4 likely form a functional hetero-tetramer. The presence of multiple copies of the related HvSINE4 element points to an intact transposition machinery of HvLINE4, despite the lack of an ORF that is equivalent to ORF1 of HvLINE2 and 3. The classification of the element, however, remains uncertain.
HmLINE1, the corresponding element to HmSINE_V2 (see above), comprises unique features, too, and is hence difficult to assign to any of the groups classified by Kojima (2019). The element contains a single ORF only that encodes a protein with, in that order, putative APE, RH and RT domains (Figure 6 and Supplementary Material Figure S9), but lacks an ORF1-like domain. Remarkably, the putative RH and RT domains have switched their positions compared to the canonical structure of LINEs (Wicker et al. 2005). Several copies of HmLINE1 are present in the genome of H. manillensis, indicating its competence for transposition.
WpLINE1 of W. pigra is a damaged element due to the integration of yet another TE, a LTR-retrotransposon, near the 5` end. The intact element very likely contained a single ORF encoding two putative APE domains as well as putative RT and RH domains. Whereas the order of domains resembles HvLINEs1-3, the distances between the domains are rather short (Figure 6 and Supplementary Material Figure S14). An unique feature of WpLINE1 is the presence of a second putative APE domain. So far, the presence of two endonuclease domains in a LINE has only been described for the elements Dualen (APE and RLE domains, Kojima and Fujiwara 2005) and Helitron (APE and HUH domains, Poulter et al. 2003). No intact copies of WpLINE1 are present in the genome of W. pigra, the element is likely non-functional.
Classification of LTR-Retrotransposons
Both WpLTR and HvLTR display remarkable structural features that make them different from all superfamilies of LTR-retrotransposons defined so far by Wicker et al. (2007) and Kojima (2019). First, they do not encode a Gag protein; second, the loss of domains in WpLTR; and third, the gain of a putative YR domain in addition to the canonical IN domain.
The Gag (or Group-specific antigen) protein comprises the retroviral matrix (MA), the capsid (CA) and the nucleocapsid (NC) proteins (Karn 2013). In LTR-retrotransposons the Gag-encoded proteins are mandatory to form a ribonucleoprotein or virus-like particle (VLP), in which the reverse transcription process takes place (Havecker et al. 2004; Sabot and Schulman 2006). Like WpLTR and HvLTR, the LTR-retrotransposon Morgane lacks Gag. But in contrast, Morgane does not encompass a functional ORF that encodes the remaining domains of LTR-retrotransposons like the RT domain. Morgane is very likely a non-autonomous TE and its transposition may hence depend on the trans-activity by a different LTR-retrotransposon protein complex (Sabot et al. 2006). The lack of Gag can indeed be compensated as described for the BARE-2 element (Tanskanen et al. 2007). The presence of several copies of WpLTR and HvLTR in the genomes of their respective hosts indicates that the elements are functional, however, the actual mode of transposition, whether autonomously or non-autonomously, remains elusive.
Based on the order of domains HvLTR belongs to either the Gypsy or the Bel-Pao, but not the Copia superfamily of LTR-retrotransposons (Wicker et al. 2007). WpLTR exhibits basically the same domain order, but lacks the RT and RH domains (Figure 6 and Supplementary Material figures S15 and S16). Strikingly, the three remaining domains of WpLTR (YR, AP and IN) can either be encoded by individual ORFs (copy 1) or by a single ORF (copies 2-5). A comparable split has also been observed for HvLINe1 and HvLINE3 compared to HvLINE2 (Figure 6).
The most striking feature of both WpLTR and HvLTR is the presence of an YR domain in addition to the canonical IN domain. The YR domain is a structural hallmark of DIRS-like elements (Goodwin and Poulter 2001; Goodwin and Poulter 2004) and the Crypton element (a DNA transposon; Goodwin et al. 2003), but is not present in any other groups of LTR-retrotransposons. DIRS1-like elements are present in a broad variety of eukaryote taxa including Annelids (Piednoël et al. 2011). YR-mediated transposition occurs via integration of a circular DNA intermediate by site-specific recombination and does not generate a TSD (Curcio and Derbyshire 2003; Poulter and Butler 2015), whereas IN-mediated transposition occurs via integration of a blunt-end DNA intermediate by a DNA cutting and joining reaction and typically generates a TSD (Nefedova and Kim 2017). The integration sites of all copies of WpLTR and HvLTR provide clear evidence for integration via an YR-mediated recombination process. The combination of structure and transposition mode justifies to define WpLTR and HvLTR as the first members of a new superfamily of LTR-retrotransposons.
Biological Significance
TEs have a deep impact on genome diversity (Warren et al. 2015), they are drivers of genome evolution (Kazazian 2004; Nishihara 2020) and may influence biological processes up to speciation (Serrato-Capuchina and Matute 2018). However, both presence and activity of TEs may also have deleterious effects on their hosts (Platt et al. 2018). The impact of TEs on leeches, however, remains speculative, mainly due to the almost complete lack of detailed information on the presence of TEs in leech genomes. The present study provides only a first glimpse of the likely diversity of TEs in leeches. However, the presence of two of these elements, namely HvSINE1 and HmSINE_V2, in hirudin genes (albeit in intron sequences) provides an excellent explanation for the remarkable redundancy of hirudin and HLF genes in leech species (Müller et al. 2016, 2017). Haematophagous leeches critically depend on the presence and activity of hirudin as a central inhibitor of blood coagulation to ensure the uptake of a blood meal (Gross and Roth 2007). A loss-of-function mutation, e.g. due to the random integration of a TE into the coding region of a hirudin gene, would certainly have an immediate negative impact on the fitness of the respective organism. Redundancy can hence be seen as strategy to compensate for putative gene losses due to such deleterious events.
Conclusions
For the first time, individual TEs have been identified and structurally characterized in leeches, a so far neglected group of animals in terms of TE research. Representatives of three types of TEs could be identified, namely SINEs, LINEs and LTR-retrotransposons, some of them unique in structure compared to canonical TEs. However, the actual diversity of TEs in leeches is likely still much higher. Non-model organisms are hence an excellent source for new information even on long-term studied objects like TEs and may provide new insights into the diversity and the putative biological impact of these fascinating genetic elements.
Acknowledgements
The author would like to thank all members of the Animal Physiology working group at the University of Greifswald for their help and support. A special thank goes to Jan-Peter Hildebrandt and Undine Lauf for critical reading of the manuscript.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Conflicts of interest
The author declares that he has no conflicts of interest.
Ethical approval
I declare that the investigations described in this paper comply with the current laws in Germany.
Data Availability Statement
Leech genome and transcriptome data for H. robusta , H. manillensis, H. medicinalis , W. pigra and H. verbana are freely accessible and searchable through public databases like GenBank. Specific sequences are provided in the Supplementary Material, further inquiries can be directed to the corresponding author.
References
- Arkhipova IR (2006) Distribution and phylogeny of Penelope-like elements in eukaryotes. Syst Biol 55(6):875-885. [CrossRef]
- Arkhipova IR (2017) Using bioinformatic and phylogenetic approaches to classify transposable elements and understand their complex evolutionary histories. Mob DNA 8:19. [CrossRef]
- Babenko VV , Podgorny OV, Manuvera VA, Kasianov AS, Manolov AI, Grafskaia EN, Shirokov DA, Kurdyumov AS, Vinogradov DV, Nikitina AS, Kovalchuk SI, Anikanov NA, Butenko IO, Pobeguts OV, Matyushkina DS, Rakitina DV, Kostryukova ES, Zgoda VH, Baskova IP, Trukhan VM, Gelfand MS, Govorun VM, Schiöth HB, Lazarev VN (2020) Draft genome sequences of Hirudo medicinalis and salivary transcriptome of three closely related medicinal leeches. BMC Genomics21(1):331. [CrossRef]
- Bell EA, Butler CL, Oliveira C, Marburger S, Yant L, Taylor MI (2022) Transposable element annotation in non-model species: The benefits of species-specific repeat libraries using semi-automated EDTA and DeepTE de novo pipelines. Mol Ecol Resour 22(2):823-833. [CrossRef]
- Borodulina OR, Kramerov DA (2001) Short interspersed elements (SINEs) from insectivores. Two classes of mammalian SINEs distinguished by A-rich tail structure. Mamm Genome 12(10):779-786. [CrossRef]
- Borodulina OR, Kramerov DA (2008) Transcripts synthesized by RNA polymerase III can be polyadenylated in an AAUAAA-dependent manner. RNA 14(9):1865-1873. [CrossRef]
- . [CrossRef]
- Borodulina OR, Golubchikova JS, Ustyantsev IG, Kramerov DA (2016) Polyadenylation of RNA transcribed from mammalian SINEs by RNA polymerase III: Complex requirements for nucleotide sequences. Biochim Biophys Acta 1859(2):355-365. [CrossRef]
- . [CrossRef]
- Bourque G, Burns KH, Gehring M, Gorbunova V, Seluanov A, Hammell M, Imbeault M, Izsvák Z, Levin HL, Macfarlan TS, Mager DL, Feschotte C (2018) Ten things you should know about transposable elements. Genome Biol 19(1):199. [CrossRef]
- Curcio MJ, Derbyshire KM (2003) The outs and ins of transposition: from mu to kangaroo. Nat Rev Mol Cell Biol 4(11):865-877. [CrossRef]
- Deragon J-M, Zhang X (2006) Short interspersed elements (SINEs) in plants: origin, classification, and use as phylogenetic markers. Syst Biol 55(6):949-956. [CrossRef]
- Dieci G, Giuliodori S, Catellani M, Percudani R, Ottonello S (2002) Intragenic promoter adaptation and facilitated RNA polymerase III recycling in the transcription of SCR1, the 7SL RNA gene of Saccharomyces cerevisiae. J Biol Chem 277(9):6903-6914. [CrossRef]
- Evgen’ev MB, Zelentsova H, Shostak N, Kozitsina M, Barskyi V, Lankenau DH, Corces VG (1997) Penelope, a new family of transposable elements and its possible role in hybrid dysgenesis in Drosophila virilis. Proc Natl Acad Sci USA 94(1):196-201. [CrossRef]
- Fillingham JS, Thing TA, Vythilingum N, Keuroghlian A, Bruno D, Golding GB, Pearlman RE (2004) A non-long terminal repeat retrotransposon family is restricted to the germ line micronucleus of the ciliated protozoan Tetrahymena thermophila. Eukaryot Cell 3(1):157-169. [CrossRef]
- . [CrossRef]
- Finnegan DJ (1989) Eukaryotic transposable elements and genome evolution. Trends Genet 5(4):103-107. [CrossRef]
- Finnegan DL (2012) Retrotransposons. Curr Biol 22(11):R432-437. [CrossRef]
- Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, Smit AF (2020) RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117(17):9451-9457. [CrossRef]
- Gazda LD, Matúz KJ, Nagy T, Mótyán JA, Tőzsér J (2020) Biochemical characterization of Ty1 retrotransposon protease. PLoS One 15(1):e0227062. [CrossRef]
- Geiduschek EP, Tocchini-Valentini GP (1988) Transcription by RNA polymerase III. Annu Rev Biochem 57:873-914. [CrossRef]
- Gilbert N, Labuda D (1999) CORE-SINEs: eukaryotic short interspersed retroposing elements with common sequence motifs. Proc Natl Acad Sci USA 96(6):2869-2874. [CrossRef]
- . [CrossRef]
- Goodwin TJD, Poulter RTM (2001) The DIRS1 group of retrotransposons. Mol Biol Evol 18(11):2067-2082. [CrossRef]
- Goodwin TJD, Butler MI, Poulter RTM (2003) Cryptons: a group of tyrosine-recombinase-encoding DNA transposons from pathogenic fungi. Microbiology (Reading) 149(Pt 11):3099-3109. [CrossRef]
- Goodwin TJD, Poulter RTM (2004) A new group of tyrosine recombinase-encoding retrotransposons. Mol Biol Evol 21(4):746-759. [CrossRef]
- Goubert C, Craig RJ, Bilat AF, Peona V, Vogan AA, Protasio AV (2022) A beginner’s guide to manual curation of transposable elements. Mob DNA 13(1):7. [CrossRef]
- Gross U, Roth M (2007) The biochemistry of leech saliva. in Michalsen et al. (Eds): Medicinal Leech Therapy, Georg Thieme Verlag KG, Stuttgart. [CrossRef]
- Guan DL, Yang J, Liu YK, Li Y, Mi D, Ma LB, Wang ZZ, Xu SQ, Qiu Q (2020) Draft Genome of the Asian buffalo leech Hirudinaria manillensis. Front Genet 10:1321. [CrossRef]
- Han G, Zhang N, Jiang H, Meng X, Qian K, Zheng Y, Xu J, Wang J (2021) Diversity of short interspersed nuclear elements (SINEs) in lepidopteran insects and evidence of horizontal SINE transfer between baculovirus and lepidopteran hosts. BMC Genomics 22(1):226. [CrossRef]
- Havecker ER, Gao X, Voytas DF (2004) The diversity of LTR retrotransposons. Genome Biol 5(6):225. [CrossRef]
- Hildebrandt J-P, Lemke S (2011) Small bite, large impact–saliva and salivary molecules in the medicinal leech, Hirudo medicinalis. Naturwissenschaften 98(12):995-1008. [CrossRef]
- Karn J (2013) Retrovirusus. In Brenner’s Encyclopedia of Genetics (2nd Ed.) Acad Press, pp211-215. [CrossRef]
- Kazazian Jr HH (2004) Mobile elements: drivers of genome evolution. Science 303(5664):1626-1632. [CrossRef]
- . [CrossRef]
- Khazina E, Weichenrieder O (2009) Non-LTR retrotransposons encode noncanonical RRM domains in their first open reading frame. Proc Natl Acad Sci USA 106(3):731-736. [CrossRef]
- . [CrossRef]
- Kojima KK, Fujiwara H (2005) An extraordinary retrotransposon family encoding dual endonucleases. Genome Res 15(8):1106-1117. [CrossRef]
- Korstian JM, Paulat NS, Platt 2nd RN, Stevens RD, Ray DA (2022) SINE-based phylogenomics reveal extensive introgression and incomplete lineage sorting in Myotis. Genes (Basel) 13(3):399. [CrossRef]
- Kramerov DA, Vassetzky NS (2011) Origin and evolution of SINEs in eukaryotic genomes. Heredity (Edinb) 107(6):487-495 . [CrossRef]
- Krishna SS, Majumdar I, Grishin NV (2003) Structural classification of zinc fingers: survey and summary. Nucleic Acids Res 31(2):532-550. [CrossRef]
- Kvist S , Manzano-Marín A, de Carle D, Trontelj P, Siddall ME (2020) Draft genome of the European medicinal leech Hirudo medicinalis (Annelida, Clitellata, Hirudiniformes) with emphasis on anticoagulants. Sci Rep 10(1):9885. [CrossRef]
- Kvist S, Utevsky S, Marrone F, Ben Ahmed R, Gajda Ł, Grosser C, Huseynov M, Jueg U, Khomenko A, Oceguera-Figueroa A, Pěsić V, Pupins M, Rouag R, Sağlam N, Świątek P, Trontelj P, Vecchioni L, Müller C (2022) Extensive sampling sheds light on species-level diversity in Palearctic Placobdella (Annelida: Clitellata: Glossiphoniiformes). Hydrobiol 849:1239-1259. [CrossRef]
- Lemke S, Vilcinskas A (2020) European Medicinal leeches - new roles in modern medicine. Biomedicines 8(5):99. [CrossRef]
- Li Y, Jiang N, Sun Y (2022) AnnoSINE: a short interspersed nuclear elements annotation tool for plant genomes. Plant Physiol 188(2):955-970. [CrossRef]
- Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR (1997) Reverse transcriptase motifs in the catalytic subunit of telomerase. Science 276(5312):561-567. [CrossRef]
- Lukas P, Melikian G, Hildebrandt J-P, Müller C (2022) Make it double: Identification and characterization of a Tandem-Hirudin from the Asian medicinal leech Hirudinaria manillensis.
- Parasitol Res 121(10):2995-3006. [CrossRef]
- Meier B, Clejan I, Liu Y, Lowden M, Gartner A, Hodgkin J, Ahmed S (2006) trt-1 is the Caenorhabditis elegans catalytic subunit of telomerase. PLoS Genet 2(2):e18. [CrossRef]
- Maris C, Dominguez C, Allain FH-T (2005) The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J 272(9):2118-2131. [CrossRef]
- Metcalfe CJ, Casane D (2014) Modular organization and reticulate evolution of the ORF1 of Jockey superfamily transposable elements. Mob DNA 5:19. [CrossRef]
- Miyamoto MM (1999) Molecular systematics: Perfect SINEs of evolutionary history? Curr Biol 9(21):R816-819. [CrossRef]
- Moelling K, Broecker F, Russo G, Sunagawa S (2017) RNase H as gene modifier, driver of evolution and antiviral defense. Front Microbiol 8:1745. [CrossRef]
- Müller C, Mescke K, Liebig S, Mahfoud H, Lemke S, Hildebrandt J-P (2016) More than just one: multiplicity of hirudins and hirudin-like factors in the medicinal leech Hirudo medicinalis.
- Mol Genet Genomics 291(1):227-240. [CrossRef]
- Müller C, Haase M, Lemke S, Hildebrandt J-P (2017) Hirudins and hirudin-like factors in Hirudinidae: implications for function and phylogenetic relationships. Parasitol Res 116(1):313-325. [CrossRef]
- Nefedova L, Kim A (2017) Mechanisms of LTR-retroelement transposition: Lessons from Drosophila melanogaster. Viruses 9(4):81. [CrossRef]
- Nikaido M, Rooney AP, Okada N (1999) Phylogenetic relationships among cetartiodactyls based on insertions of short and long interpersed elements: hippopotamuses are the closest extant relatives of whales. Proc Natl Acad Sci USA 96(18):10261-10266. [CrossRef]
- Nishihara H (2020) Transposable elements as genetic accelerators of evolution: contribution to genome size, gene regulatory network rewiring and morphological innovation. Genes Genet Syst 94(6):269-281. [CrossRef]
- Ohta S, Tsuchida K, Choi S, Sekine Y, Shiga Y, Ohtsubo E (2002) Presence of a characteristic D-D-E motif in IS1 transposase. J Bacteriol 184(22):6146-6154. [CrossRef]
- Okada N, Hamada M, Ogiwara I, Ohshima K (1997) SINEs and LINEs share common 3’ sequences: a review. Gene 205(1-2):229-243. [CrossRef]
- Paulsen RT, Agany DDM, Petersen J, Davis CM, Ehli EA, Gnimpieba E, Burrell BS (submitted 2020, version 2023) A draft genome for Hirudo verbana, the Medicinal leech. [CrossRef]
- Piednoël M, Gonçalves IR, Higuet D, Bonnivard E (2011) Eukaryote DIRS1-like retrotransposons: an overview. BMC Genomics 12:621. [CrossRef]
- Platt 2nd RN, Vandewege MW, Ray DA (2018) Mammalian transposable elements and their impacts on genome evolution. Chromosome Res 26(1-2):25-43. [CrossRef]
- Poulter RTM, Goodwin TJD, Butler MI (2003) Vertebrate helentrons and other novel Helitrons. Gene 313:201-212. [CrossRef]
- Poulter RTM, Goodwin TJD (2005) DIRS-1 and the other tyrosine recombinase retrotransposons. Cytogenet Genome Res 110(1-4):575-588. [CrossRef]
- Poulter RTM, Butler MI (2015) Tyrosine recombinase retrotransposons and transposons. Microbiol Spectr 3(2):MDNA3-0036-2014. [CrossRef]
- Quesneville H (2020) Twenty years of transposable element analysis in the Arabidopsis thaliana genome. Mob DNA 11:28. eCollection 2020. [CrossRef]
- Ray DA, Xing J, Salem A-H, Batzer MA (2006) SINEs of a nearly perfect character. Syst Biol 55(6):928-935. [CrossRef]
- Roy-Engel AM (2012) A tale of an A-tail: The lifeline of a SINE. Mob Genet Elements 2(6):282-286. [CrossRef]
- Sabot F, Schulman AH (2006) Parasitism and the retrotransposon life cycle in plants: a hitchhiker’s guide to the genome. Heredity (Edinb) 97(6):381-388. [CrossRef]
- Sabot F, Sourdille P, Chantret N, Bernard M (2006) Morgane, a new LTR retrotransposon group, and its subfamilies in wheats. Genetica 128(1-3):439-447. [CrossRef]
- SenGupta D (2013) RNA-Binding Domains in Proteins. In Brenner’s Encyclopedia of Genetics (2nd Ed.) Acad Press pp274-276. [CrossRef]
- Serrato-Capuchina A, Matute DR (2018) The role of transposable elements in speciation. Genes (Basel) 9(5):254. [CrossRef]
- Shi J, Liang C (2019) Generic Repeat Finder: A high-sensitivity tool for genome-wide de novo repeat detection. Plant Physiol 180(4):1803-1815. [CrossRef]
- Simakov O, Marletaz F, Cho S-J, Edsinger-Gonzales E, Havlak P, Hellsten U, Kuo D-H, Larsson T, Lv J, Arendt D, Savage R, Osoegawa K, de Jong P, Grimwood J, Chapman JA, Shapiro H, Aerts A, Otillar RP, Terry AY, Boore JL, Grigoriev IV, Lindberg DR, Seaver EC, Weisblat DA, Putnam NH, Rokhsar DS (2013) Insights into bilaterian evolution from three spiralian genomes. Nature 493(7433):526-531. [CrossRef]
- Tanskanen JA, Sabot F, Vicient C, Schulman AH (2007) Life without GAG: the BARE-2 retrotransposon as a parasite’s parasite. Gene 390(1-2):166-174. [CrossRef]
- Tarailo-Graovac M, Chen N (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4:Unit 4.10. [CrossRef]
- Tözsér J (2010) Comparative studies on retroviral proteases: substrate specificity. Viruses 2(1):147-165. [CrossRef]
- Tong L, Dai S-X, Kong D-J, Yang P-P, Tong X, Tong X-R, Bi X-X, Su Y, Zhao Y-Q, Liu Z-C (2022) The genome of medicinal leech (Whitmania pigra) and comparative genomic study for exploration of bioactive ingredients. BMC Genomics 23(1):76. [CrossRef]
- Traboni C, Ciliberto G, Cortese R (1982) A novel method for site-directed mutagenesis: its application to an eukaryotic tRNAPro gene promoter. EMBO J 1(4):415-420. [CrossRef]
- Trontelj P, Utevsky SY (2012) Phylogeny and phylogeography of medicinal leeches (genus Hirudo): fast dispersal and shallow genetic structure. Mol Phylogenet Evol 63(2):475-485. [CrossRef]
- Warren IA, Naville M, Chalopin D, Levin P, Berger CS, Galiana D, Volff J-N (2015) Evolutionary impact of transposable elements on genomic diversity and lineage-specific innovation in vertebrates. Chromosome Res 23(3):505-531. [CrossRef]
- Wells JN, Feschotte C (2020) A field guide to eukaryotic transposable elements. Annu Rev Genet 54:539-561. [CrossRef]
- Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8(12):973-982. [CrossRef]
- Wu S, Zhang X, Han J (2016) A computational model for predicting RNase H domain of retrovirus. PLoS One 11(8):e0161913. [CrossRef]
- Zhao F, Huang Z, He B, Liu K, Li J, Liu Z, Lin G (2024) Comparative genomics of two Asian medicinal leeches Hirudo nipponia and Hirudo tianjinensis: With emphasis on antithrombotic genes and their corresponding proteins. Int J Biol Macromol 270(Pt 1):132278. [CrossRef]
Figure 1.
Schematic representation of hirudin HV1 gene structures of Hirudo verbana. Exons are labeled in green, introns in red and the putative short interspersed nuclear element HvSINE1 in light blue. The integration site of HvSINE1 is indicated. Sizes are adjusted relative to the size of exon1. HGW: biological sample used for the analyses in Müller et al. (2017); USA: biological sample used for the analyses in Paulsen et al. (2020, version 2023).
Figure 1.
Schematic representation of hirudin HV1 gene structures of Hirudo verbana. Exons are labeled in green, introns in red and the putative short interspersed nuclear element HvSINE1 in light blue. The integration site of HvSINE1 is indicated. Sizes are adjusted relative to the size of exon1. HGW: biological sample used for the analyses in Müller et al. (2017); USA: biological sample used for the analyses in Paulsen et al. (2020, version 2023).
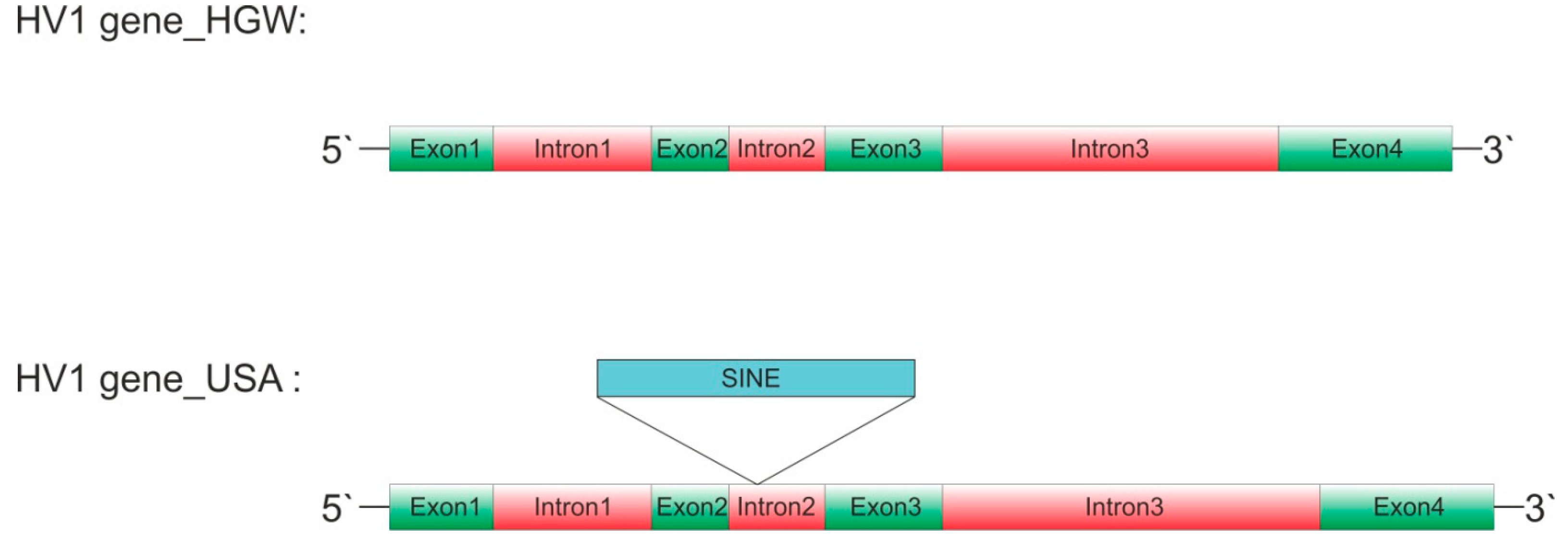
Figure 2.
Consensus sequences for promoter regions of the 5S rRNA gene (A), tRNA and 7SL genes (B) and the respective tRNA gene of Hirudo verbana and Hirudo medicinals and predicted HvSINE1-4 promoters (C). +Dieci et al. (2002); *Geiduschek and Tocchini-Valentini (1988); # Traboni et al. (1982) Taken together, the putative TE very likely represents a SINE and was hence termed HvSINE1.
Figure 2.
Consensus sequences for promoter regions of the 5S rRNA gene (A), tRNA and 7SL genes (B) and the respective tRNA gene of Hirudo verbana and Hirudo medicinals and predicted HvSINE1-4 promoters (C). +Dieci et al. (2002); *Geiduschek and Tocchini-Valentini (1988); # Traboni et al. (1982) Taken together, the putative TE very likely represents a SINE and was hence termed HvSINE1.

Figure 3.
Sequences of putative SINEs of Hirudo verbana. Predicted tRNA-gene derived promoter regions (Box1 and Box2) are marked in bold. The conserved central domain is underlined. Simple repeat regions in HvSINE4 are labeled in yellow and red. The LINE-specific region of HvSINE4 is double underlined.
Figure 3.
Sequences of putative SINEs of Hirudo verbana. Predicted tRNA-gene derived promoter regions (Box1 and Box2) are marked in bold. The conserved central domain is underlined. Simple repeat regions in HvSINE4 are labeled in yellow and red. The LINE-specific region of HvSINE4 is double underlined.

Figure 4.
Schematic representation of phylogenetic trees based on HvSINE1-like element (left) and coi (right) sequences.
Figure 4.
Schematic representation of phylogenetic trees based on HvSINE1-like element (left) and coi (right) sequences.
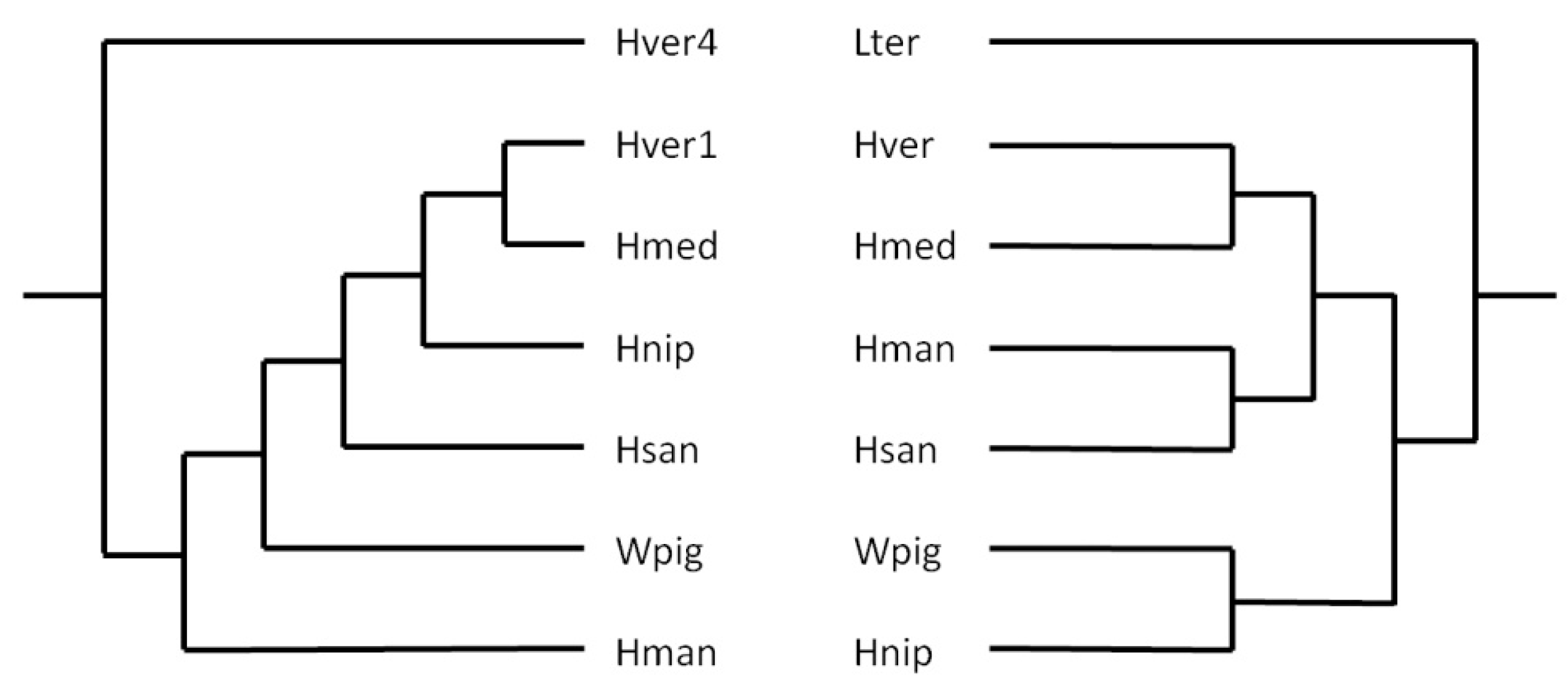
Figure 5.
Sequence of HmSINE_V2 of Hirudinaria manillensis. The predicted tRNA-gene derived promoter region (Box1 and Box2), the pol III terminator sequence and the pAS are marked in bold. The sequence stretch that results in BLAST search hits with genome sequences of H. verbana, H. medicinalis and W. pigra is underlined.
Figure 5.
Sequence of HmSINE_V2 of Hirudinaria manillensis. The predicted tRNA-gene derived promoter region (Box1 and Box2), the pol III terminator sequence and the pAS are marked in bold. The sequence stretch that results in BLAST search hits with genome sequences of H. verbana, H. medicinalis and W. pigra is underlined.

Figure 6.
Schematic drawings of putative long interspersed nuclear elements (LINEs) of H. verbana (HvLINE1-4), H. manillensis (HmLINE1) and W. pigra (WpLINE1) and of long terminal repeat (LTR) elements of W. pigra and H. verbana. Size and orientation of open reading frames are indicated by open arrows. Colored marks indicate the position of characteristic sequence motifs like the CCHC motif (green), the RRM motif (dark and light blue), a polyA motif (orange), putative TSDs (red) and LTRs (purple)
Figure 6.
Schematic drawings of putative long interspersed nuclear elements (LINEs) of H. verbana (HvLINE1-4), H. manillensis (HmLINE1) and W. pigra (WpLINE1) and of long terminal repeat (LTR) elements of W. pigra and H. verbana. Size and orientation of open reading frames are indicated by open arrows. Colored marks indicate the position of characteristic sequence motifs like the CCHC motif (green), the RRM motif (dark and light blue), a polyA motif (orange), putative TSDs (red) and LTRs (purple)


Table 1.
Comparison of HV1 gene structures of Hirudo verbana and Hirudo medicinalis.
| Gene | Exon1 | Intron1 | Exon2 | Intron2 | Exon3 | Intron3 | Exon4 |
| Hv_HGW1 | 61 | 103 | 50 | 62 | 76 | 199 | 71 |
| Hv_HGW2 | 61 | 103 | 50 | 62 | 76 | 199 | 71 |
| Hv_USA | 61 | 103 | 50 | 272 | 76 | 224 | 71 |
| Hm_HGW1 | 61 | 103 | 50 | 62 | 76 | 199 | 71 |
| Hm_HGW2 | 61 | 103 | 50 | 62 | 76 | 199 | 71 |
| Hm_Kvist | 61 | 103 | 50 | 62 | 76 | 199 | 71 |
Red boxes indicate the prominent differences in size of exon2 and exon3. Source of sequence data: Hv_HGW: Müller et al. (2017); Hm_HGW: Müller et al. (2016); Hm_Kvist: Kvist et al. (2020); Hv_USA: Paulsen et al. (2020, version 2023).
Table 2.
Abundance of short interspersed nuclear elements HvSINEs in genomes of H. verbana and H. medicinalis. For HvSINE4, 21/14 copies contain the whole sequence, whereas about 200 copies contain the head and core domain, but lack the LINE-specific segment.
Table 2.
Abundance of short interspersed nuclear elements HvSINEs in genomes of H. verbana and H. medicinalis. For HvSINE4, 21/14 copies contain the whole sequence, whereas about 200 copies contain the head and core domain, but lack the LINE-specific segment.
| Hirudo verbana | Hirudo medicinalis | |
| HvSINE1: | > 1000 copies | > 1000 copies |
| HvSINE2: | 1 copy | 1 copy |
| HvSINE3: | 1 copy | 1 copy |
| HvSINE4: | 21 (about 200) copies | 14 (about 200) copies |
Table 3.
Expression pattern of HvSINE-RNAs in Hirudo verbana and/or Hirudo medicinalis tissues.
| HvSINE1 | HvSINE2 | HvSINE3 | HvSINE4 | |
| salivary gland | ✓ | - | - | ✓ |
| muscle | ✓ | - | - | ✓ |
| ganglion | ✓ | ✓ | - | ✓ |
| CNS | ✓ | ✓ | ✓ | ✓ |
| head | ✓ | ✓ | ✓ | ✓ |
Table 4.
Presence of HvSINE1-like sequences in genomes of other Annelids.
| Hirudinaria manillensis | + |
| Whitmania pigra | + |
| Hirudo nipponia | + |
| Haemopis sanguisuga | + |
| Limnobdella mexicana | - |
| Macrobdella decora | - |
| Asiaticobdella fenestrata | - |
| Haemadipsa interrupta | - |
| Haementeria vizzotoi | - |
| Helobdella robusta | - |
| Piscicola geometra | - |
| Enchytraeus crypticus | - |
| Eisenia fetida | |
| Capitella teleta |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.


Epigenetic Regulation of A Heat-Activated Retrotransposon in Cruciferous Vegetables
Kosuke Nozawa
et al.
,
2017
Diversity and Evolution of pogo and Tc1/mariner Transposons in the Apoidea Genome
Yibing Liu
et al.
,
2021



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated