
Preprint
Article
The First Homoleptic Geometrical Anti-zn Complex from an Unsymmetric Curcuminoid
Altmetrics
Downloads
87
Views
55
Comments
0
A peer-reviewed article of this preprint also exists.


This version is not peer-reviewed
Submitted:
12 August 2024
Posted:
12 August 2024
You are already at the latest version
Alerts
Abstract
Curcuminoids are widely studied due to their well-recognized therapeutic properties. These molecules are often derivatized with metals, rendering their corresponding homoleptic metal complexes. Numerous crystal structures of homoleptic symmetric curcuminoids with physiologically essential metals are known, although the literature lacks reports of homoleptic unsymmetric metal complexes of curcuminoids (or hemi-curcuminoids). Two unknowns must be solved when an unsymmetric curcuminoid ligand is reacted with a metal ion: a) the degree of coordination (MLn) and b) the conformational nature (syn or anti) of the complex. Herein, we report the structure of the anti-isomer of the Zn complex of the hemi-curcuminoid 5-hydroxy-1-(4-methoxyphenyl)hexa-1,4-dien-3-one. While the NMR shows only one set of signals for this homoleptic complex, the unambiguous stereochemistry was established through single-crystal X-ray diffractometry, revealing a hexacoordinated ML2 structure.

Keywords:
Subject: Chemistry and Materials Science - Medicinal Chemistry
1. Introduction
Curcumin, the main active metabolite [1] of the spice Curcuma sp., has garnered scientific interest due to its potential in treatment of Alzheimer's disease. Its therapeutic properties as anti-cancer, antioxidant, and anti-inflammatory [2], offer a promising avenue for research in biochemistry and pharmacology. Structurally, curcumin's beta-diketone group is in equilibrium of two possible tautomers in solution (keto and enol [3]); it also comprises two α,β-unsaturated systems [4] and a chain of seven carbon atoms [5] flanked by two aromatic rings substituted with para-hydroxy (-OH) and meta-methoxy (-OCH3) groups (see Figure 1).
The synthetic derivation of curcumin and the preparation of analogous compounds gives rise to different families of compounds called curcuminoids, such as diaryl-heptanoids [6], hemi-curcuminoids [7] (monoaryl-hexanoids), monocarbonyl curcuminoids [8,9] (diaryl-pentanoids) or half-curcuminoids [10](monoaryl-propanoids) and are exemplified in Figure 2.
After the derivatization of phenolic groups to methoxy or acetyl groups, the diarylheptanoids dimethoxy-curcumin and diacetyl curcumin (DAC) are obtained. However, when the double bonds are hydrogenated [11], the curcuminoids (e.g. tetrahydrocurcumin or hexahydrocurcumin) are obtained but still are considered part of the diarylheptanoid family.
It is important to note that curcuminoids are commonly derivatized with metals aiming to overcome inherent problems such as reduced aqueous solubility and low bioavailability [12,13]. Structural studies of curcuminoids with transition metals (e.g. Mg or Zn) have established stoichiometric ratios of 1:1 or 1:2. It is known that the two types of metal complexes that have been reported in the literature are homoleptic and heteroleptic complexes with symmetrical structural characteristics. Heteroleptic curcuminoid complexes [14] occur when complexation involves different ligands (e.g., bipyridines or phenanthrolines [15]), while homoleptic complexes arise when the same curcuminoid ligand occupies all the complexation sites (see Figure 3).
Analogue compounds inspired by the skeleton of curcumin [16] are named hemi-curcuminoids [17]. They can be obtained synthetically by preserving the skeleton's structural half, i.e., an aromatic ring, the beta-diketone system, and the α,β-unsaturated function. A characteristic of this type of compound is its unsymmetrical nature [16], involving different molecular fragments at each side of the β-diketone function (see Figure 2). These compounds containing the β -diketone function can be readily deprotonated, giving rise to enolates capable of chelating with different metal ions. Although the metal coordination chemistry of the symmetric curcuminoid family has received much attention, the synthesis of unsymmetric curcuminoid metal complexes is unexplored with few examples in the literature [5]. In the latter case, the possible syn or anti-isomerism resulting from this new type of metal complex must be assessed. In addition, unsymmetric curcuminoids are not readily available, and synthesis using the commonly described methods leads also to symmetric curcuminoids [18].
Zinc is an element involved in various biochemical processes [19] and is considered crucial in healthy metabolism. However, zinc metal curcuminoid complexes are biologically active against different cancer cell lines, and their physicochemical properties (e.g., aqueous solubility) exceed those of their parent curcuminoid ligands.
Zinc complexes are interesting in coordination chemistry due to their structural variety and diverse geometries [20] observed when a ligand (symmetric or unsymmetric) is reacted with such a metal ion. In addition, the degree of coordination (MLn) and the unambiguous geometry are best answered when established by X-ray of the single crystal technique. Several zinc homoleptic complexes of curcuminoids have been authenticated by single crystal studies [21,22], with the following geometries: square pyramidal, trigonal pyramidal, trigonal bipyramidal, or octahedral. In all these cases, symmetric curcuminoid ligands were used, and the solvent of crystallization plays an important role in defining the geometry, as shown in Figure 4.
Herein, we report the preparation of an unsymmetric ligand (hemicurcuminoid type) using a new synthetic approach. Thus, a mono-ketalization reaction of 2,4-pentanedione with ethanediol, followed by the aldol mono condensation reaction with anisaldehyde in alkaline media, and further hydrolysis of the mono-ketal affords the target compound containing the beta diketone function.
Finally, the synthetic unsymmetric curcuminoid (5-hydroxy-1-(4-methoxyphenyl)hexa-1,4-dien-3-one) was reacted with zinc acetate and a suitable single crystal was obtained successfully by slow evaporation in methanol which provides three important structural features: a) metal-ligand relationship (MLn), b) coordination geometry, and c) the syn or anti isomerism (the latter being a new structural aspect for this type of complexes). All compounds were fully characterized using spectroscopic techniques.
2. Materials and Methods
Acetylacetone (acac, 2,4-pentanodione), ethanediol, p-toluenesulfonic acid, 4-methoxybenzaldehyde (anisaldehyde, CAS 123-11-5), zinc acetate, high-purity grade silica gel, average pore size 60 Å (52–73 Å), 70–230 mesh (CAS 112926-00-8), and all solvents HPLC grade were purchased from Sigma-Aldrich and were used without prior purification.
Melting points were obtained in an Electrothermal Engineering IA9100 digital melting point apparatus in open capillary tubes and were uncorrected [23]. 1H, 13C NMR spectra were obtained in a Bruker Fourier 400 MHz spectrometer using TMS as an internal reference and DMSO-d6 or CDCl3 as solvent. NMR spectra were processed with Mestre Nova software 12.0.3-21384 [24] and are found in the Supplementary Materials. IR absorption spectra were recorded using an FT-IR NICOLET IS-50, Thermo Fisher Scientific spectrophotometer in the range of 4000-400 cm-1 with a reflectance technique using an ATR diamond accessory [25]. Mass Spectra were recorded using The MStation JMS-700 JEOL equipment (Electron Ionization impact positive mode), the AccuTOF JMS-T100LC JEOL equipment (DART+, positive ion mode) and a Bruker Esquire 6000 equipment [26] (ESI-TI, APCI-TI).
Single-crystal X-ray Diffraction. The Rigaku Diffraction Xcalibur Atlas, Gemini CCD diffractometer was used for a single-crystal X-ray diffraction analysis of Compound 4, with a graphite monochromator and MoKα source of radiation (λ = 0.71073 Å) by a ω scan at 130K temperature. The collection and reduction of data were performed by the CrysAlis software package [27]. Crystal structure was solved using direct methods by SHELXS [28] and refined by the SHELXL [29] program. The structures were refined by the full-matrix least-squares method based on F2 against all reflections. All non-hydrogen atoms were refined anisotropically. The hydrogen atom of the methanol ligand was found in the Fourier difference map, and its positional parameters were refined. The methoxy phenyl moiety is disordered and was modeled over two positions, with occupancies refined to about 0.5. Structures were visualized by MERCURY [30], and the geometrical parameters were calculated by PLATON [31]. The crystallographic data for Compound 4 are summarized in Supplementary Materials.
Preparation of Compound 1 (Mono-ketal).
In a 250 mL round flask provided with a Dean-Stark apparatus, 5 mL of 2,4-pentanodione (50 mmol) was dissolved in 50 mL of benzene, then 1.4 mL of ethanediol (23 mmol) was added, after that 10 mg of p-toluenesulfonic acid monohydrate was added, and the reaction was left in reflux for 3 hours. Finally, after the removal of the solvent, the title product was purified by a reduced pressure distillation (50° C and 5 mm Hg).
Reaction of mono-condensation for obtained of compound 2.
In a 100 ml round flask, 1.7 mL of 4-methoxybenzaldehyde (14 mmol, anisaldehyde) was dissolved in 50 mL of methanol; then 2 g of compound 1 (12 mmol) dissolved in methanol was added dropwise. Later 0.5 g de NaOH (12.5 mmol) finely powdered was added to the vessel. The reaction was left with magnetic stirring at room temperature for 3 days. The solvent was evaporated in vacuo and an extraction with water (100 mL) and ethyl acetate (100 mL) 1:1 was carried out. The organic phase was dried with Na2SO4 and concentrated. The product was purified by SiO2 column chromatography using a 6:4 hexane/ ethyl acetate mixture as eluent.
Synthesis of compound 3 (Unsymmetric hemicurcuminoid, enol type).
In a 100 mL round flask, 1.05 g of compound 2 was dissolved in 40 mL of methanol. Further, 2 mL of HCl (4.6 mmol) was added and the reaction was conducted with magnetic stirring at room temperature for 6 hours. The solvent was evaporated and the residue was extracted with a sodium bicarbonate saturated solution (50 mL) and ethyl acetate (50 mL) 1:1; the organic phase was dried with Na2SO4 and concentrated.
Synthesis of unsymmetric homoleptic complex and preparation of single crystal.
In a 100 mL round flask, 0.2 g of compound 3 was dissolved in 10 mL of ethyl acetate. Subsequently, 0.085 g of zinc acetate (4.6 mmol) dissolved in methanol were added dropwise. The reaction was left with magnetic stirring at room temperature for 3 hours and the precipitate was filtered off on a Hirsh funnel; the powder was washed with distilled water and dried in vacuum. 30 mg of the complex was dissolved in methanol and left standing for slow evaporation for 24 hours in the dark.
Compound 1. 1-(2-methyl-1,3-dioxolan-2-yl)propan-2-one, yield 60%. Colorless liquid. 1H NMR (300 MHz, CDCl3) δ 3.98 (m, 4H), 2.78 (s, 2H), 2.22 (s, 3H), 1.41 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 206.00, 107.82, 64.61, 52.52, 31.58, 24.37. IR-ATR 2985 cm-1, 2886 cm-1, 1707 cm-1, 1183 cm-1, 1047 cm-1. DART+-MS: m/z = [M+H]+ 145.
Compound 2. Yield 40%. Oily brown liquid. (E)-4-(4-methoxyphenyl)-1-(2-methyl-1,3-dioxolan-2-yl)but-3-en-2-one. 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 16.0 Hz, 1H), 7.51 (d, J = 8.7 Hz, 2H), 6.91 (m, 2H), 6.76 (d, J = 16.0 Hz, 1H), 3.98 (m, 4H), 3.84 (s, 3H), 2.98 (s, 2H), 1.46 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 196.85, 161.78, 143.07, 130.30, 127.41, 124.72, 114.54, 108.56, 64.88, 55.54, 50.64, 24.97. IR 1677 cm-1, 1644 cm-1, 1592 cm-1, 1510 cm-1. DART+-MS: m/z = [M+H]+ 263.
Compound 3. (1E,4Z)-5-hydroxy-1-(4-methoxyphenyl)hexa-1,4-dien-3-one. Yield 50%. Crystalline yellow solid. Melting point 68.5°C. 1H NMR (400 MHz, CDCl3) δ 15.48 (s, 1H), 7.55 (d, J = 15.8 Hz, 1H), 7.46 (m, 2H), 6.89 (m, 2H), 6.33 (d, J = 15.8 Hz, 1H), 5.61 (s, 1H), 3.83 (s, 3H), 2.14 (s, 3H).13C NMR (100 MHz, CDCl3) δ 197.15, 178.09, 161.29, 139.75, 129.69, 127.93, 120.51, 114.50, 100.86, 55.49, 26.95. IR-ATR 1629 cm-1, 1282 cm-1, 1109 cm-1. IE+-MS: m/z = [M]+ 218.
Compound 4. Zinc Complex of 5-hydroxy-1-(4-methoxyphenyl)hexa-1,4-dien-3-one acetate. Yield 75%. Yellow powder. Melting point 94°C. 1H NMR (400 MHz, DMSO-d6) δ 7.59 (m, 4H), 7.38 (d, J = 15.7 Hz, 2H), 6.95 (m, 4H), 6.59 (d, J = 15.7 Hz, 2H), 5.48 (s, 2H), 3.78 (s, 6H), 2.08 (s, 6H), 1.96 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 206.50, 193.27, 181.93, 160.21, 137.03, 129.31, 128.04, 127.00, 114.32, 100.63, 55.25, 30.68, 28.28. IR-ATR 1632 cm-1, 1602 cm-1, 1505 cm-1, 450 cm-1. ESI+-MS: m/z = 521.4 [M + Na]+.
3. Results
The production of a target hemi-curcuminoid compound (enol type) was achieved through the aldol mono-condensation reaction of 2,4-pentanedione (ACAC). Since there are two reactive terminal carbons at both ends susceptible to condensation, ACAC was functionalized blocking by ketalization one of the carbonyl groups with 1,2-ethanediol in an acidic medium (mono-ketal synthesis, see Figure 5). The structure of the mono-ketal from 2,4-pentanedione was determined spectroscopically. Thus, the proton magnetic resonance [32] (1H NMR) showed multiple signals near 4 ppm assigned to the AA´BB´ system corresponding to compound 1 (see Supplementary Materials). In addition, the mass spectrometry revealed a prominent peak at m/z = 145 for the molecular ion that corresponds adequately to the formula C7H12O3.
The adequate reactivity of the terminal methyl of compound 1 allows the aldol mono-condensation reaction with anisaldehyde in basic media (see Figure 6). The 1H NMR signals of compound 2 showed two doublets at d 7.54 ppm (b) and 6.76 ppm (a) assigned to the α,β-unsaturated vinyl system with coupling constants J =16 Hz, correlating appropriately for the trans configuration. Additionally, the proton spectrum (see Supplementary Materials) showed multiple signals at d 4 ppm due to the ketal group. The mass spectrum of compound 2 showed a peak at m/z = 263 [M+H]+ which corresponds to the molecular formula C15H18O4
The opening of the mono-ketal (compound 2) in an acidic medium is illustrated in Figure 6 affording hemicurcuminoid 3 (recovery the keto-enol system). The 1H NMR spectrum of compound 3 shows two simple signals characteristic of the enol, i.e., the hydroxyl (-OH) was observed at 15. 48 ppm (hydrogen bond) [33] and the methine (-CH-) at 5.61 ppm. In addition, the vinyl system characteristic for this compound is confirmed by the presence of two double signals at d 7.55 ppm (β) and d 6.33 ppm (α) with couplings trans J = 16 Hz [34] (see Supplementary Materials). Compound 3 has a molecular formula C13H14O3 and was verified by a peak in the mass spectrum at m/z = 218 [M]+, which is expected for the molecular ion.
In principle, the reaction of the hemicurcuminoid with zinc acetate can lead to two conformational isomers, i.e., syn and anti as illustrated in Figure 7. The liquid state NMR data helped in the characterization of the complex but were not conclusive regarding the authentication of the molecular geometry. Therefore, the detailed single crystal X-ray analysis was imperative to answer this question.
Proton magnetic resonance was indicative of the presence of the metal ion in the complex. When comparing the chemical shifts of the pure ligand with those of the zinc complex, a shift towards lower frequencies is observed [22] (Table 1). Surprisingly, compound 4 showed only one set of hydrogen and carbon-13 signals (see Supplementary Materials), which is indicative of the presence of a single isomer in solution (DMSO-d6).
4. Discussion
The most desirable and common trend in the synthesis of curcuminoid-derived in the form of metal complexes is to find new biologically active compounds that respond to the needs of human ailments such as inflammation, cancer, and Alzheimer's disease. However, the architectural design of such relatively simple compounds may require an unambiguos structural determination before any biological testing is performed. In this order of ideas, the core of this research is to answer the structural unknowns of a new zinc complex (compound 4) that is obtained with an unsymmetric curcuminoid ligand.
The nuclear magnetic resonance spectrum for compound 4 showed signals corresponding to a single isomer and is the first time that a resonance spectrum of an unsymmetric curcuminoid ligand with zinc has been shown (see Figure 8 and Supplementary Materials). Finding a single set of NMR signals is unexpected because other zinc homoleptic complexes have shown two or more sets of NMR signals [35], thus the explanation will be in conjunction with X-rays. The NMR spectrum of compound 4 shows a singlet at 2.08 ppm (see Supplementary Materials) which corresponds to the acetate group coordinated with zinc. Upon crystallization of the crude precipitate in methanol, these acetate groups are replaced by methanol molecules as revealed by the x-ray crystal structure. Furthermore, the integration of signals normalized to two protons for the methine group (-CH at 5.48 ppm) allowed us to propose an ML2-type hexacoordinated complex [22]. Although the geometry could not be unambiguously determined until the X-rays were available, the new complex of zinc was clearly of homoleptic type.
The work of Dawid Jędrzkiewicz et.al. [35], found that the zinc complex (L2Zn) with an unsymmetric ligand (named O-dtBu, N-C12) produced more than one set of signals for the H-NMR spectrum which is attributed to a mixture of isomers in solution (syn-dimer and anti-dimer). The single set of signals observed in the proton spectrum of compound 4 in Figure 8, could correspond in principle to the syn or anti-isomer, but remarkably only one isomer is observed in solution.
As it can be appreciated in Figure 9, both possible structures syn and anti, have the symmetry elements C2 [36], and both would show a complete overlap of signals with possible differences in chemical shifts due to anisotropic effects from aromatic rings. Therefore, the optimal way to assess the geometry and configuration of the complex is X-ray crystallography.
The anti-isomer too has the C2 rotation axis with n = 180° and a symmetry plane (σv) that produces its reflection (shown in Figure 9), so these elements could explain the production of magnetically equivalent signals for each ligand around zinc. It is also interesting to note that an unsymmetric curcuminoid ligand produces magnetically equivalent signals when spatially arranged in the anti-form.
The synthesis and characterization of transition metal complexes of hemi-curcuminoids are scarce. There is only a couple of studies in which the monoaryl-hexanoids appear as a ligand in metal complexes [37,38], and a survey using the CSD (Version 5.45, update of June 2024) [39] revealed only three structure determinations (Ref Codes: JELZAF, JELZIN and JELZEJ) all of them for heteroleptic Ru(η6-p-cymene) complexes of three hispolon derivatives. To the best of our knowledge, this constitutes the synthesis and chemical characterization of the first homoleptic anti-Zn complex from an unsymmetric curcuminoid.
The asymmetric unit of compound 4 consists of one half of the neutral complex with th Zn having Ci symmetry lying on an inversion center, one deprotonated (1E,4Z)-5-hydroxy-1-(4-methoxyphenyl)hexa-1,4-dien-3-one molecule (Compound 3) in the equatorial plane and one coordinated methanol molecule in apical position (Figure 10). The coordination geometry of compound 4 corresponds to a slightly apical distorted octahedron, presumed by Jahn–Teller effect, with zinc–oxygen from the coordinated methanol in apical/axial positions (2.201(3) longer than the equatorial zinc–oxygen bonds from the ligand (2.010(2)— 2.070(2)Å), all these values are slightly longer than zinc–oxygen bonds (equatorial: 1.986(3)–2.037(5) Å, axial 2.252(6) Å) of the close related compound B showed in Figure 4.
In the complex, the deprotonated (1E,4Z)-5-hydroxy-1-(4-methoxyphenyl)hexa-1,4-dien-3-one ligand displays a fully extended conformation with a significantly deviation of planarity. Two planes can be observed corresponding to the 5-hydroxy-hexa-1,4-dien-3-one moiety (Rms deviation = 0.0212 Å ) and the 4-methoxyphenyl group (Rms deviation of fitted atoms = 0.0395 Å), making a dihedral angle of 28.85(0.18)°. A similar trend is observed in the heteroleptic Ru(η6-p-cymene) complexes of three hispolon derivatives (JELZAF: 28.01°, JELZIN: 14.32° and JELZEJ: 4.93°) revealing the high flexibility of this type of ligands. In the crystal complex molecules are held together by hydrogen bonds O4-H4A...O1(x-1, y, z): 2.10(5) Å, forming dimers, Figure 11.
5. Conclusions
This research has established the guidelines for a structural problem of isomerism in unsymmetric curcuminoid metal complexes. It is also crucial to recognize the value of complementary single-crystal X-ray structural analyses for curcuminoid-derived metal complexes. It is important to note that an apparent simple molecular design demands detailed structural studies before carrying out a series of biological assays.
To the best of our knowledge, this report constitutes the first crystal structure of a metal complex of an unsymmetric curcuminoid. Although the structure found has anti-configuration, it remains unknown if the syn configuration is also present at some stage of the complex formation. X-ray crystallography offered the most conclusive answer regarding the preferred geometry of the complex. A detailed study of the thermodynamics associated with the complex formation may provide insight into determining the selectivity of the preferred geometric isomer.
Supplementary Materials
The deposit number CCDC 2376717 (Compound 4) contains the supplementary crystallographic data for this article, including structure factors. These data can be obtained free of charge at https://www.ccdc.cam.ac.uk/structures/ (accessed date 09 August 2024). The following supporting information can be downloaded at: Preprints.org.
Author Contributions
Conceptualization, R.G.E.; methodology, M.A.O.M, G.M.E., R.T.H., R.S.O; software, M.A.O.M, G.M.E., R.T.H., R.S.O., R.A.T.; validation, R.G.E, and R.A.T; formal analysis, M.A.O.M, G.M.E., R.T.H., R.S.O., R.A.T., R.G.E.; investigation, M.A.O.M, G.M.E., R.T.H., R.S.O.; resources, R.G.E.; data curation, R.G.E, and R.A.T.; writing—original draft preparation, R.G.E.; writing—review and editing, R.G.E.; visualization, R.G.E.; supervision, R.G.E.; project administration, R.G.E.; funding acquisition, R.G.E.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by CONAHCyT, grant number FOINS-PRONACES-307152 and PAPIIT DGAPA-UNAM, grant number IT200720.
Data Availability Statement
Not applicable.
Acknowledgments
Raúl G. Enríquez acknowledges support from CONAHCyT (FOINS-PRONACES-307152) and DGAPA-UNAM (IT200720). Marco A. Obregón-Mendoza acknowledges honorary payment from CONAHCyT (FOINS-PRONACES-307152). R.T.H. acknowledges CONAHCyT by the postdoctoral fellowship (CVU-662794) and G.M.E. CVU-1347017 to CONAHCyT scholarship. Acknowledgments are extended to Elizabeth Huerta (NMR), Isabel Chávez (NMR), Adriana Romo (IR), María del Carmen García (MS), Eréndira García Ríos (ESI) and Lucero Ríos (ESI) from Instituto de Química-UNAM.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jiang, Y.; Du, Z.; Xue, G.; Chen, Q.; Lu, Y.; Zheng, X.; Conney, A.H.; Zhang, K. Synthesis and Biological Evaluation of Unsymmetrical Curcumin Analogues as Tyrosinase Inhibitors. Molecules 2013, 18, 3948–3961. [CrossRef]
- Saladini, M.; Lazzari, S.; Pignedoli, F.; Rosa, R.; Spagnolo, F.; Ferrari, E. New Synthetic Glucosyl-Curcuminoids, and Their 1H and 13C NMR Characterization, from Curcuma Longa L. Plant Foods for Human Nutrition 2009, 64, 224–229. [CrossRef]
- Torres-Rodríguez, E.; Arias-Cedeño, Q.; Almeida-Saavedra, M.; Michalik-Michalik, M.; Vogel-Vogel, C. Study of the Keto-Enolic Equilibrium in Structures of Synthetic Curcuminoids by Means of RMN and X Rays Diffraction. Revista Cubana de Química 2013, XXV, 206.
- Haritakun, W.; Changtam, C. Cytotoxic Activity of Curcuminoids and Curcuminoid Analogues Against Human Oral Cancer KB Cells. SDU Res. J 2016, 9, 141–158.
- Deepthi, T. V.; Venugopalan, P. Synthesis, DNA-Binding, and Cytotoxic Studies on Three Copper(II) Complexes of Unsymmetrical Synthetic Analogues of Curcumin. J Coord Chem 2016, 69, 3403–3416. [CrossRef]
- Singh, R.; Tønnesen, H.H.; Vogensen, S.B.; Loftsson, T.; Másson, M. Studies of Curcumin and Curcuminoids. XXXVI. The Stoichiometry and Complexation Constants of Cyclodextrin Complexes as Determined by the Phase-Solubility Method and UV-Vis Titration. J Incl Phenom Macrocycl Chem 2010, 66, 335–348. [CrossRef]
- Cornago, P.; Cabildo, P.; Sanz, D.; Claramunt, R.M.; Torralba, M.C.; Torres, M.R.; Elguero, J. Structures of Hemi-Curcuminoids in the Solid State and in Solution. European J Org Chem 2013, 6043–6054. [CrossRef]
- Shetty, D.; Kim, Y.J.; Shim, H.; Snyder, J.P. Eliminating the Heart from the Curcumin Molecule: Monocarbonyl Curcumin Mimics (MACs). Molecules 2015, 20, 249–292.
- Dong, L.; Zheng, S.; Zhang, Y.; Jiang, X.; Wu, J.; Zhang, X.; Shan, X.; Liang, D.; Ying, S.; Feng, J.; et al. Design, Synthesis, and Evaluation of Semi-Conservative Mono-Carbonyl Analogs of Curcumin as Anti-Inflammatory Agents against Lipopolysaccharide-Induced Acute Lung Injury. Medchemcomm 2015, 6, 1544–1553. [CrossRef]
- Obregón-Mendoza, M.A.; Arias-Olguín, I.I.; Meza-Morales, W.; Alvarez-Ricardo, Y.; Chávez, M.I.; Toscano, R.A.; Cassani, J.; Enríquez, R.G. Expected and Unexpected Products in Half Curcuminoid Synthesis: Crystal Structures of but-3-En-2-Ones and 3-Methylcyclohex-2-Enones. Crystals (Basel) 2021, 11. [CrossRef]
- Masuda, T.; Matsumura, H.; Oyama, Y.; Takeda, Y.; Jitoe, A.; Kida, A.; Hidaka, K. Synthesis of (+/-)-Cassumunins A and B, New Curcuminoid Antioxidants Having Protective Activity of the Living Cell against Oxidative Damage. J Nat Prod 1998, 61, 609–613.
- Wanninger, S.; Lorenz, V.; Subhan, A.; Edelmann, F.T. Metal Complexes of Curcumin - Synthetic Strategies, Structures and Medicinal Applications. Chem Soc Rev 2015, 44, 4986–5002. [CrossRef]
- Prasad, S.; Dubourdieu, D.; Srivastava, A.; Kumar, P.; Lall, R. Metal–Curcumin Complexes in Therapeutics: An Approach to Enhance Pharmacological Effects of Curcumin. Int J Mol Sci 2021, 22. [CrossRef]
- Mittal, A.; Nagpal, M.; Vashistha, V.K.; Arora, R.; Issar, U. Recent Advances in the Antioxidant Activity of Metal-Curcumin Complexes: A Combined Computational and Experimental Review. Free Radic Res 2024, 58, 11–26. [CrossRef]
- Figueroa-Depaz, Y.; Pérez-Villanueva, J.; Soria-Arteche, O.; Martínez-Otero, D.; Gómez-Vidales, V.; Ortiz-Frade, L.; Ruiz-Azuara, L. Casiopeinas of Third Generations: Synthesis, Characterization, Cytotoxic Activity and Structure–Activity Relationships of Mixed Chelate Compounds with Bioactive Secondary Ligands. Molecules 2022, 27. [CrossRef]
- Raduly, F.M.; Raditoiu, V.; Raditoiu, A.; Grapin, M.; Fierascu, R.C.; Raut, I.; Constantin, M. Functionalized Palygorskite as a Delivery Platforms for Bioactive Asymmetric Beta-Diketone Dyes. Crystals (Basel) 2024, 14, 659. [CrossRef]
- Zimnitskiy, N.S.; Korotaev, V.Y.; Barkov, A.Y.; Kochnev, I.A.; Sosnovskikh, V.Y. Hemicurcuminoids (1-Styryl-1,3-Diketones) - Valuable Multi-Faceted Building Blocks for Organic Synthesis. New Journal of Chemistry 2023, 47, 5110–5149.
- Cheng, Y.J.; Li, C.W.; Kuo, C.L.; Shih, T.L.; Chen, J.J. Improved Synthesis of Asymmetric Curcuminoids and Their Assessment as Antioxidants. Molecules 2022, 27. [CrossRef]
- Maywald, M.; Rink, L. Zinc Deficiency and Zinc Supplementation in Allergic Diseases. Biomolecules 2024, 14, 863. [CrossRef]
- Bharti, A.; Bharati, P.; Chaudhari, U.K.; Singh, A.; Kushawaha, S.K.; Singh, N.K.; Bharty, M.K. Syntheses, Crystal Structures and Photoluminescent Properties of New Homoleptic and Heteroleptic Zinc(II) Dithiocarbamato Complexes. Polyhedron 2015, 85, 712–719. [CrossRef]
- Meza-Morales, W.; Mirian Estévez-Carmona, M.; Alvarez-Ricardo, Y.; Obregón-Mendoza, M.A.; Cassani, J.; Ramírez-Apan, M.T.; Escobedo-Martínez, C.; Soriano-García, M.; Reynolds, W.F.; Enríquez, R.G. Full Structural Characterization of Homoleptic Complexes of Diacetylcurcumin with Mg, Zn, Cu, and Mn: Cisplatin-Level Cytotoxicity in Vitro with Minimal Acute Toxicity in Vivo. Molecules 2019, 24. [CrossRef]
- Meza-Morales, W.; Alvarez-Ricardo, Y.; Obregón-Mendoza, M.A.; Arenaza-Corona, A.; Ramírez-Apan, M.T.; Toscano, R.A.; Poveda-Jaramillo, J.C.; Enríquez, R.G. Three New Coordination Geometries of Homoleptic Zn Complexes of Curcuminoids and Their High Antiproliferative Potential. RSC Adv 2023, 13, 8577–8585. [CrossRef]
- Obregón-Mendoza, M.A.; Estévez-Carmona, M.M.; Alvarez-Ricardo, Y.; Meza-Morales, W.; Escobedo-Martínez, C.; Soriano-García, M.; Enríquez, R.G. Crystal Structure, Synthesis and Biological Activity of Ether and Ester Trans-Ferulic Acid Derivatives. Int J Org Chem (Irvine) 2018, 8, 359–377. [CrossRef]
- Mestrelab Research. MNova Software. Available Online Https://Mestrelab.Com/Download/Mnova/ (Accesed on March, 10, 2024).
- Obregón-Mendoza, M.A.; Meza-Morales, W.; Alvarez-Ricardo, Y.; Estévez-Carmona, M.M.; Enríquez, R.G. High Yield Synthesis of Curcumin and Symmetric Curcuminoids: A “Click” and “Unclick” Chemistry Approach. Molecules 2023, 28. [CrossRef]
- Arenaza-Corona, A.; Obregón-Mendoza, M.A.; Meza-Morales, W.; Ramírez-Apan, M.T.; Nieto-Camacho, A.; Toscano, R.A.; Pérez-González, L.L.; Sánchez-Obregón, R.; Enríquez, R.G. The Homoleptic Curcumin–Copper Single Crystal (ML2): A Long Awaited Breakthrough in the Field of Curcumin Metal Complexes. Molecules 2023, 28. [CrossRef]
- Rigaku Oxford Diffraction: Yarnton, U. CrysAlisPRO Software System 2024.
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr A 2008, 64, 112–122. [CrossRef]
- Sheldrick, G.M. SHELXT - Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr A 2015, 71, 3–8. [CrossRef]
- MacRae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J Appl Crystallogr 2020, 53, 226–235. [CrossRef]
- Spek, A.L. Single-Crystal Structure Validation with the Program PLATON. J. Appl. Cryst 2003, 36, 7–13.
- Coffin, A.; Ready, J.M. Selective Synthesis of (+)-Dysoline. Org Lett 2019, 21, 648–651. [CrossRef]
- Zawadiak, J.; Mrzyczek, M. Correlation of Substituted Aromatic β-Diketones’ Characteristic Protons Chemical Shifts with Hammett Substituent Constants. Magnetic Resonance in Chemistry 2013, 51, 689–694. [CrossRef]
- Li, W.; Wang, S.; Feng, J.; Xiao, Y.; Xue, X.; Zhang, H.; Wang, Y.; Liang, X. Structure Elucidation and NMR Assignments for Curcuminoids from the Rhizomes of Curcuma Longa. Magnetic Resonance in Chemistry 2009, 47, 902–908. [CrossRef]
- Jȩdrzkiewicz, D.; Marszałek-Harych, A.; Ejfler, J. Serendipitous Synthesis Found in the Nuances of Homoleptic Zinc Complex Formation. Inorg Chem 2018, 57, 8169–8180. [CrossRef]
- Rachwalski, M. Special Issue: Asymmetry and Symmetry in Organic Chemistry. Symmetry (Basel) 2023, 15.
- Wei, X.; Yang, Y.; Ge, J.; Lin, X.; Liu, D.; Wang, S.; Zhang, J.; Zhou, G.; Li, S. Synthesis, Characterization, DNA/BSA Interactions and in Vitro Cytotoxicity Study of Palladium(II) Complexes of Hispolon Derivatives. J Inorg Biochem 2020, 202. [CrossRef]
- Caruso, F.; Subbaraju, G. V.; Ramani, M. V.; Gariboldi, M.; Marras, E.; Kloer, C.; Sulovari, A.; Kaur, S.; Rossi, M. Synthesis, X-Ray Diffraction and Anti-Proliferative Biological Activity of Hispolon Derivatives and Their (H6-p-Cymene)(Hispolonato)Ruthenium[II] Chloride Complexes. Inorganica Chim Acta 2022, 542. [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr B Struct Sci Cryst Eng Mater 2016, 72, 171–179. [CrossRef]
Figure 1.
Curcumin structure.

Figure 2.
Families of curcuminoids.

Figure 3.
Type of metal complexes from curcumin, M= metal ion.

Figure 4.
Several geometries of homoleptic zinc complexes, A: square pyramidal (CCDC 1453160), B: octahedral (CCDC 2234961) [22], C: trigonal pyramid (CCDC 2234962) and D: distorted trigonal-bipyramidal (CCDC 2234963), hydrogens have been omitted.
Figure 4.
Several geometries of homoleptic zinc complexes, A: square pyramidal (CCDC 1453160), B: octahedral (CCDC 2234961) [22], C: trigonal pyramid (CCDC 2234962) and D: distorted trigonal-bipyramidal (CCDC 2234963), hydrogens have been omitted.

Figure 5.
Preparation of mono-ketal from 2,4-pentanodione.

Figure 6.
Synthesis of unsymmetric hemicurcuminoid from anisaldehyde.
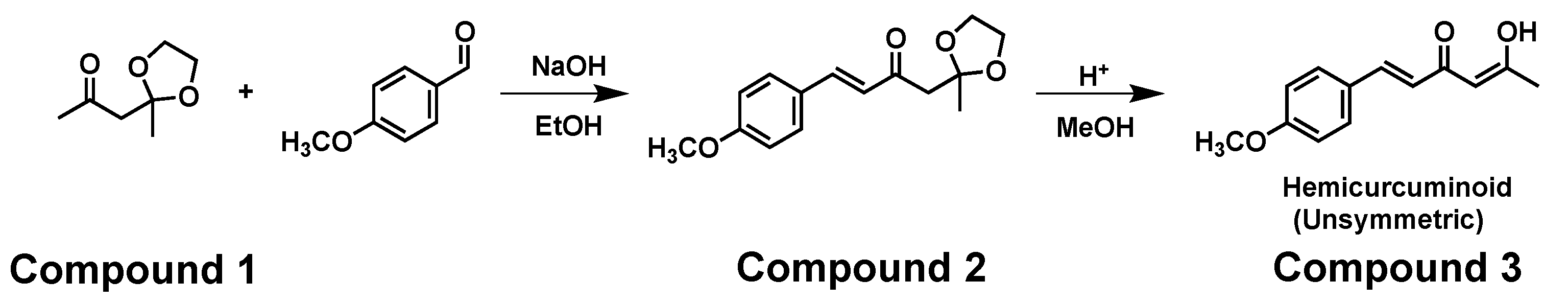
Figure 7.
Synthesis of unsymmetric metal complex.

Figure 8.
1H NMR spectra of hemicurcuminoid and their zinc complex (400 MHz, DMSO-d6).

Figure 9.
Symmetry operations for syn and anti-isomer complexes.

Figure 10.
ORTEP-like drawing and coordination geometry of Compound 4, 50% probability level of thermal ellipsoids, unlabeled atoms generated by inversion centre symmetry (minor disordered 4-methoxyphenyl atoms are omitted for clarity).
Figure 10.
ORTEP-like drawing and coordination geometry of Compound 4, 50% probability level of thermal ellipsoids, unlabeled atoms generated by inversion centre symmetry (minor disordered 4-methoxyphenyl atoms are omitted for clarity).

Figure 11.
Crystal structure of Compound 4 along the b-axis. Hydrogen bonds represented by broken line.
Figure 11.
Crystal structure of Compound 4 along the b-axis. Hydrogen bonds represented by broken line.


Table 1.
Chemical Shifts of ligand (compound 3) and their zinc complex (compound 4).
| Hydrogen | Compound 3, δ = ppm 1 | Compound 4, δ = ppm1 |
|---|---|---|
| -CH3 | 2.13 (singlet) | 1.96 (singlet) |
| =C-H (α) | 6.66 (doublet) | 6.59 (doublet) |
| =C-H (β) | 7.54 (doublet) | 7.38 (doublet) |
| -CH (Methine) | 5.87 (singlet) | 5.48 (singlet) |
| Ar-H (AA´) | 6.99 (multiplet) | 6.95 (multiplet) |
| Ar-H (BB´) | 7.64 (multiplet) | 7.59 (multiplet) |
| -O-CH3 | 3.80 (singlet) | 3.78 (singlet) |
| -OH (enol) | 15.65 (broad) | - |
| -OOC-CH3 (acetate) | - | 2.08 (singlet) |
1 Solvent DMSO-d6.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.


Synthesis, Structure, DFT and In Silico Studies of Zn (II) Complex with Lipoic Acid: A Potential Antidiabetic Drug
Farkhod Jumabaev
et al.
,
2023
Synthesis and Cytotoxicity Evaluation of Novel Asymmetrical Mono-carbonyl Analogs of Curcumin (AMACs) against Vero, HeLa, and MCF7 Cell Lines
Pekik Wiji Prasetyaningrum
et al.
,
2018
Combining a Drug and a Nutraceutical: A New Cocrystal of Praziquantel and Curcumin
Camila Caro Garrido
et al.
,
2024



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated